
 ACCESS
Research Article
ACCESS
Research Article
Volume 2, Article ID: 2025.0009

Mohammad Reza Shakibaie
mr_shakibaei@kmu.ac.ir


Samane Shakibaie
samaneshakibaie@gmail.com

1 Department of Microbiology and Virology, Kerman University of Medical Sciences, Kerman 7616913555, Iran Tel/fax: +983433257665
2 Medical Informatics Research Center, Kerman University of Medical Sciences, Kerman 7616913555, Iran
3 Research Center of Tropical and Infectious Diseases, Kerman University of Medical Sciences, Kerman 7616913555, Iran
4 Medical Mycology and Bacteriology Research Center, Kerman University of Medical Sciences, Kerman 7616913555, Iran
* Author to whom correspondence should be addressed
Received: 26 Apr 2025 Accepted: 13 Jun 2025 Available Online: 13 Jun 2025 Published: 27 Jun 2025
Limited information is currently available regarding the contraction mechanism of the Hcp-1 hemolysin-coregulated protein within the type VI secretion system (T6SS) of Acinetobacter baumannii, particularly about the secretion of toxic effectors. This research aimed to evaluate the mechanism underlying the contraction of the Hcp1 nanotube in response to the putative toxic effector TagX protein by employing bioinformatics tools and the artificial intelligence (AI) DeepMind system. To achieve this goal, the amino acid sequence of Hcp1 from A. baumannii was retrieved from the GenBank database under accession no. WP_000653195.1 and its three-dimensional (3D) structure as a whole tail and monomer was generated utilizing the AlphaFold2 AI from DeepMind. The 3D structure of the TagX hydrolase from A. baylyi ADP1 was predicted by the ExPASy auto-modeling software (ExPASy). The prepared TagX ligand was subsequently docked to the target site of selected amino acids within the Hcp1 nanotube via AutoDock Vina in the Schrödinger platform. The conformational changes of the amino acid side chains upon docking were examined using RoseTTAFold, and side chain rotational angles (Ψ/Φ) were detected based on the Dunbrack rotamer package. The molecular docking indicated that TagX toxin binds to His108, Arg98, Ser78, and Glu121 within the Hcp1 nanotube by H-bond. This interaction induces a rotameric shift in the dihedral angles of the aforementioned amino acids, initiating the contraction of the Hcp1 tail/tube via VgrG1 (gp27) contractile sheath and ClpV ATPase. This resulted in the propulsion of the effector molecule through Hcp1 to the tip of the T6SS complex and subsequent injection into prey cells.
Gram-negative bacteria thrive in competitive environments by secreting a putative toxic effector that can kill neighboring cells [1]. They have evolved various secretory mechanisms such as the type VI secretion system (T6SS) to survive harsh conditions and eliminate competitors in mixed environments while protecting themselves and their sister cells from inhibitory action of various effector toxins with complex cognate immunity genes (antidote), typically located upstream or downstream of the corresponding T6SS toxic effector gene [2]. T6SS, a macromolecular nanomachine, functions like a syringe and needle, injecting effectors (proteins) into neighboring cells. This process, occurring via cell-to-cell contact, is a mechanism used by bacteria to deliver toxins, compete with other microbes, and even interact with eukaryotic cells [3]. A. baumannii possesses a type VI secretion system which is used to kill competing bacteria, but the mechanisms and regulatory systems of its effectors are not fully understood. Particularly, the specific functions of T6SS-dependent effectors within target cells and the regulatory systems that control T6SS expression in A. baumannii require further investigation [4,5]. The Type VI Secretion System is a bacterial weapon that allows cells to compete with each other by delivering toxins into other cells. This advantage is especially pronounced in natural environments like soil, where microbial communities are diverse and resource competition is intense. Bacteria secrete various putative effector molecules—including nucleases, amidases, phospholipases, peptidoglycan hydrolases, neuraminidases, glycosidases, DNase, and ADP-ribosyl transferases—and deliver these cytotoxins into prey cells through direct cell-to-cell contact [6,7]. The Hcp and valine-glycine repeat protein G (VgrG) are considered the hallmarks of this secretion system, because they are secreted into the extracellular environment, thereby can be detected in the supernatants of all bacteria with a functional T6SS [8]. The crystal structure of Hcp1 protein in A. baumannii has been delineated by X-ray crystallography [9]. It has a tail-like tube configuration, composed of 13 putative homohexameric rings stacked to form a 600 nm-long nanotube, which functions both as a virulence factor and plays a role in biofilm formation [9]. Recent structural analysis of the Hcp1 in A. baumannii by X-ray crystallography revealed a tight β-barrel formed by two β-sheets flanked at one side by a short α-helix, and in most cases, six Hcp molecules associate to form a donut-shaped hexamer [10]. The T6SS needle structure, used to deliver proteins into target cells, is assembled on a baseplate. The inner Hcp tube, which is composed of Hcp proteins, polymerizes and is wrapped by a contractile sheath composed of TssB and TssC proteins. This sheath, when it contracts, propels the effector to the tip of T6SS by a spike of VgrG and PAAR proteins, through the baseplate and into a neighboring cell. The VgrG protein, and particularly the VgrG-PAAR spike, plays a crucial role in both assembly and the delivery of the effector proteins [11]. Studies of the Hcp1 proteins in Acinetobacter species using bioinformatics and X-ray tomography techniques showed that C-terminal extension toxins (Hcp-ET) indeed exert antibacterial properties and suppress the target cell growth when the putative toxic effector binds to the inner surface [12]. T6SS-positive A. baumannii shows unique clinical characteristics such as increased virulence of bacteria, along with a greater biofilm formation through an increase in expression of quorum sensing genes compared to T6SS-negative strains [13]. Similarly, T6SS-positive strains have a higher mortality rate in animal models, and increased Hcp expression correlates well with A. baumannii invasiveness during respiratory infections, potentially induced by acidic conditions [14]. Furthermore, Hcp1 protein was detected in the sera of cystic fibrosis patients during Burkholderia mallei infection in humans or horses and functions as a molecular chaperone [15]. A recent report revealed that interaction of the effector with the putative Hcp pore is a general requirement for secretion of diverse toxic effectors encompassing several putative enzymatic classes. Electron microscopy analysis of an Hcp–effector complex from P. aeruginosa revealed the effector bound to the inner surface of Hcp and transferred through Hcp pore to reach the needle-like structure on the tip of the T6SS [16]. It is noteworthy to mention bacterial species have adapted their T6SS to specific hosts, environments, or survival strategies. For example, the TagX peptidoglycan hydrolase from A. baylyi showed homology to known bacteriophage l, d-endopeptidases, and its amino acid sequence was conserved in the T6SS clusters of several bacterial species [17]. This study aims to investigate the structure of the Hcp1 protein in both its individual and homohexameric forms, focusing on the conformational changes in Hcp1’s ring structure when interacting with toxic effector molecules in A. baumannii.
2.1. Analysis of 3D Structure of Hcp1 Monomer To analyze the structural and functional characteristics of the Hcp1 model, the amino acid sequence (accession number WP_000653195.1) from Acinetobacter (multispecies) was retrieved from the GenBank (NCBI) database. This sequence was used as a query to identify the preliminary structure, arrangement of α-helices and β-sheets, protein folding, expected positional error, and degree of homology with similar proteins through a BLAST search. The sequence was then submitted to various bioinformatics and AI-based platforms, including SWISS-MODEL (http://www.expasy.org/), UniProtKB (https://www.uniprot.org/), a deep learning software RoseTTAFold through CAMEO (https://robetta.bakerlab.org/model), and AlphaFold2 DeepMind (https://alphafold.ebi.ac.uk/). The sequence alignment was performed in the UniProtKB database and compared with at least 5 most closely related sequences. Similarly, the secondary structure of the Hcp1 protein monomer model was predicted in such a way to generate the inner surface that binds to effector molecules, achieving an accuracy exceeding 90 in terms of the predicted local distance difference test (pLDDT) and predicted aligned error (PAE) values plot (http://www.subtiwiki.uni-goettingen.de/v4/pae). Finally, the quality controls for the accuracy of the predicted models were assessed using Z-score, RMSD index, B-factor, and MolProbity using the Ramachandran plot. The visualization of the T6SS component structures was carried out by PyMOL through the Schrödinger platform (https://www.schrodinger.com/products/pymol). The high-quality model was then selected for further experiments. 2.2. Generating the Putative TagX Toxic Effector Molecule as Ligand The putative toxic effector molecule employed here as a template was the peptidoglycan L-alanyl-D-glutamate Cwlk hydrolase (TagX) from A. baylyi ADP1. The amino acid sequence of the TagX protein was already reported [18] and was used to construct a three-dimensional structure of this toxic effector using the ExPASy auto modeling server (https://swissmodel.expasy.org/interactive). 2.3. Molecular Docking Analysis and Virtual Screening The putative TagX toxic effector protein of A. baylyi ADP1 was used as a ligand molecule, and four amino acids involved in binding to the effector within the Hcp1 ring were considered as the receptor. Molecular docking was performed using AutoDock vina in the Schrödinger platform. Protein-ligand interaction complexes were visualized using PyMOL to identify binding positions of active site residues and their binding distances with the Schrödinger module. The best docking poses were selected directly using the Glide by the Force field-based. The H-bonds were formed between the ligand molecule and four amino acids within the Hcp1 homohexamer. This is required to minimize the free energy. Here, the free energy (ΔG) was minimized using GROMACS software (http://www.gromacs.org/ /2024.0/) to create an index file with preferred dihedral angles. 2.4. Determination of Hcp1 Physicochemical Parameters To evaluate the predictive properties of Hcp1 investigated in this study, we used the ProtParam software version 2.0.5 (https://web.expasy.org/protparam/). ProtParam is a computational tool that facilitates the calculation of various physical and chemical parameters for user-entered protein sequences. The parameters computed include molecular weight, theoretical isoelectric point (pI), amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and the grand average of hydropathicity (GRAVY). 2.5. Kabsch Algorithm and the PAE Plot The goal of the Kabsch algorithm is to determine the optimal rotation matrix R that minimizes the root-mean-square deviation between two sets of points. Nevertheless, the algorithm operates in three steps: translation, computation of the covariance matrix, and rotation. Further methodological details are available at: https://pymolwiki.org/index.php/Kabsch site. Meanwhile, the PAE is useful for assessing confidence between domains or chains. 2.6. Rotamer Library An important part of this study was to determine how the toxic effector is propelled from the inner membrane to the outer membrane injectosome (VgrG1 + PAAR+ Hcp1). To answer this question, the open-source software package rotag was used to create a rotameric library using the https://www.github.com/agrybauskas/rotag site. Rotag is a collection of tools that generate and analyze side-chain rotamer libraries from protein structure data. Here, to confirm the rotameric shifts or dihedral angles of the staggered amino acids within the Hcp1 monohexameric ring, we used rotamer libraries (rotag). Rotameric libraries were used to predict amino acid dihedral angle (Φ/ψ) side chains and backbone (χ1/χ2 and Cγ) for the possibility of rotation and protein conformation. The effect of backbone conformation on side-chain rotamer is primarily due to steric repulsion between backbone atoms Cγ, whose position depends on φ and ψ side-chain heavy atoms, and is detected by the Ramachandran plot. One of the greatest advantages of rotag is its ability to deal with nonstandard amino acids. Likewise, the current rotameric library and residue data are available at the Dunbrack website (http://dunbrack.fccc.edu/bbdep/). 2.7. Statistical Analysis All analyses were performed using SPSS, version 20 (SPSS Inc., Chicago, IL, USA). The p values were two-tailed; p ≤ 0.01 was considered statistically significant. Means and standard deviations (SD) were calculated as required for numerical variables, or unpaired t-tests were used for statistical comparisons.
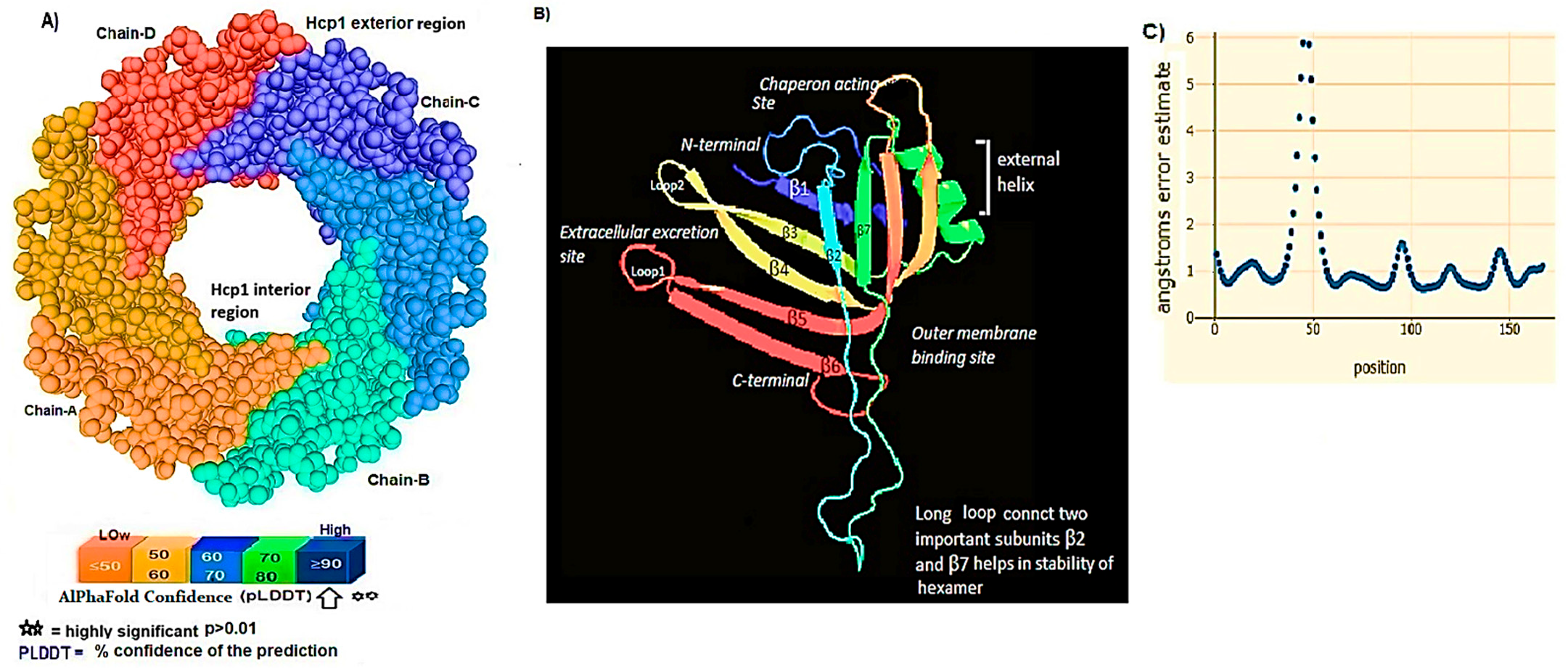
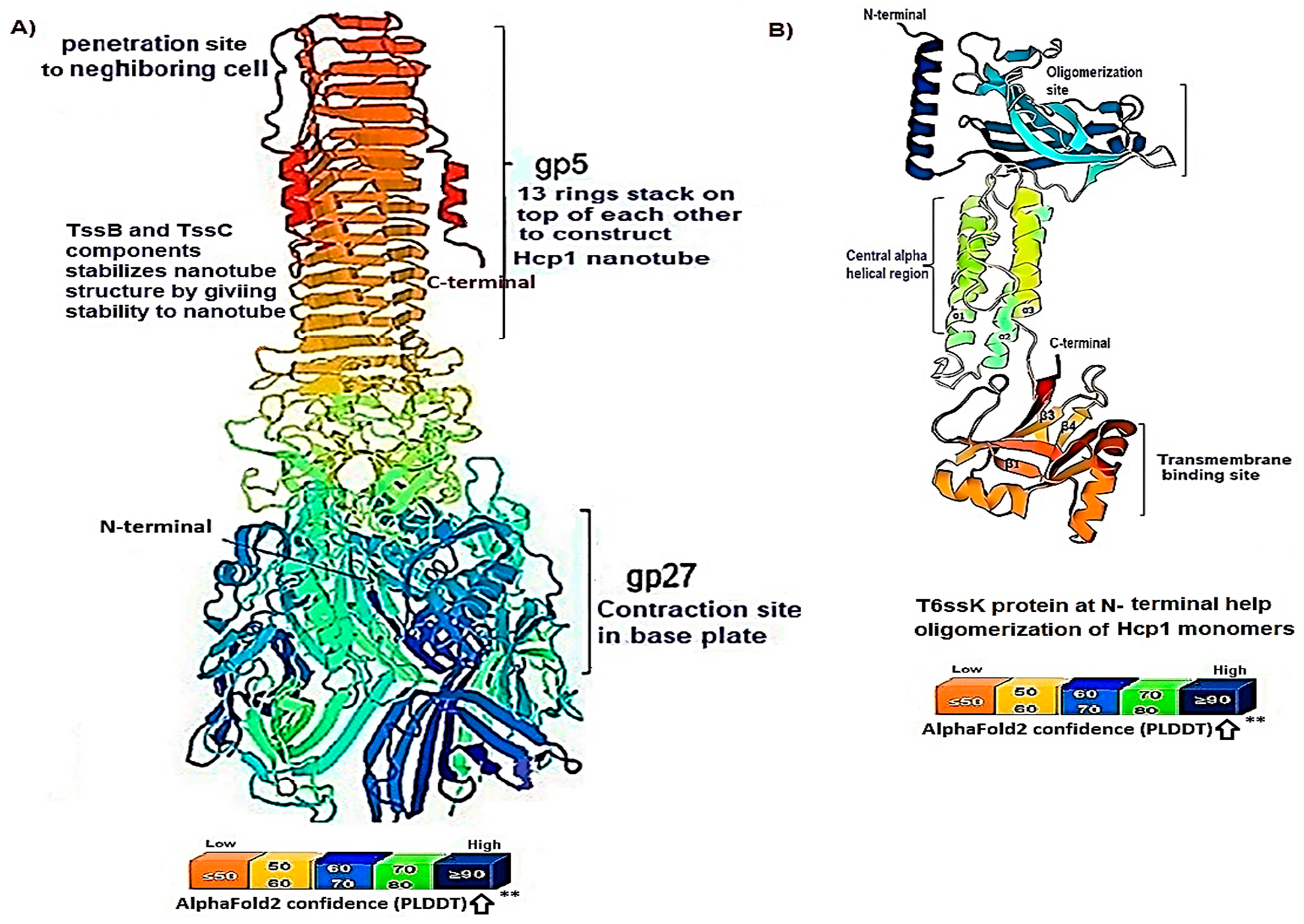
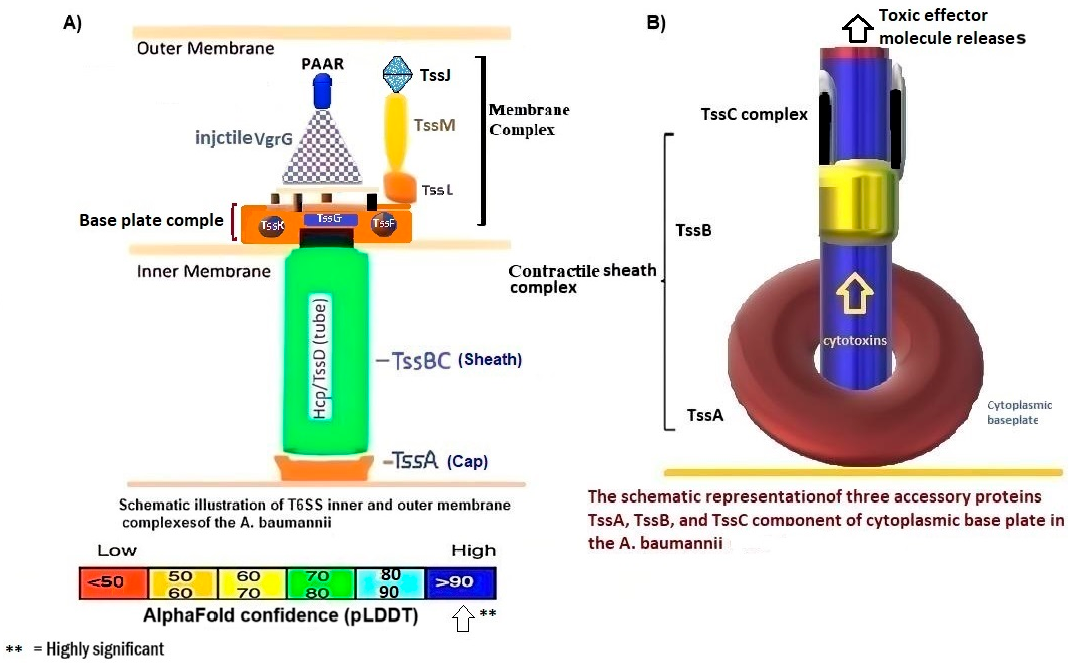
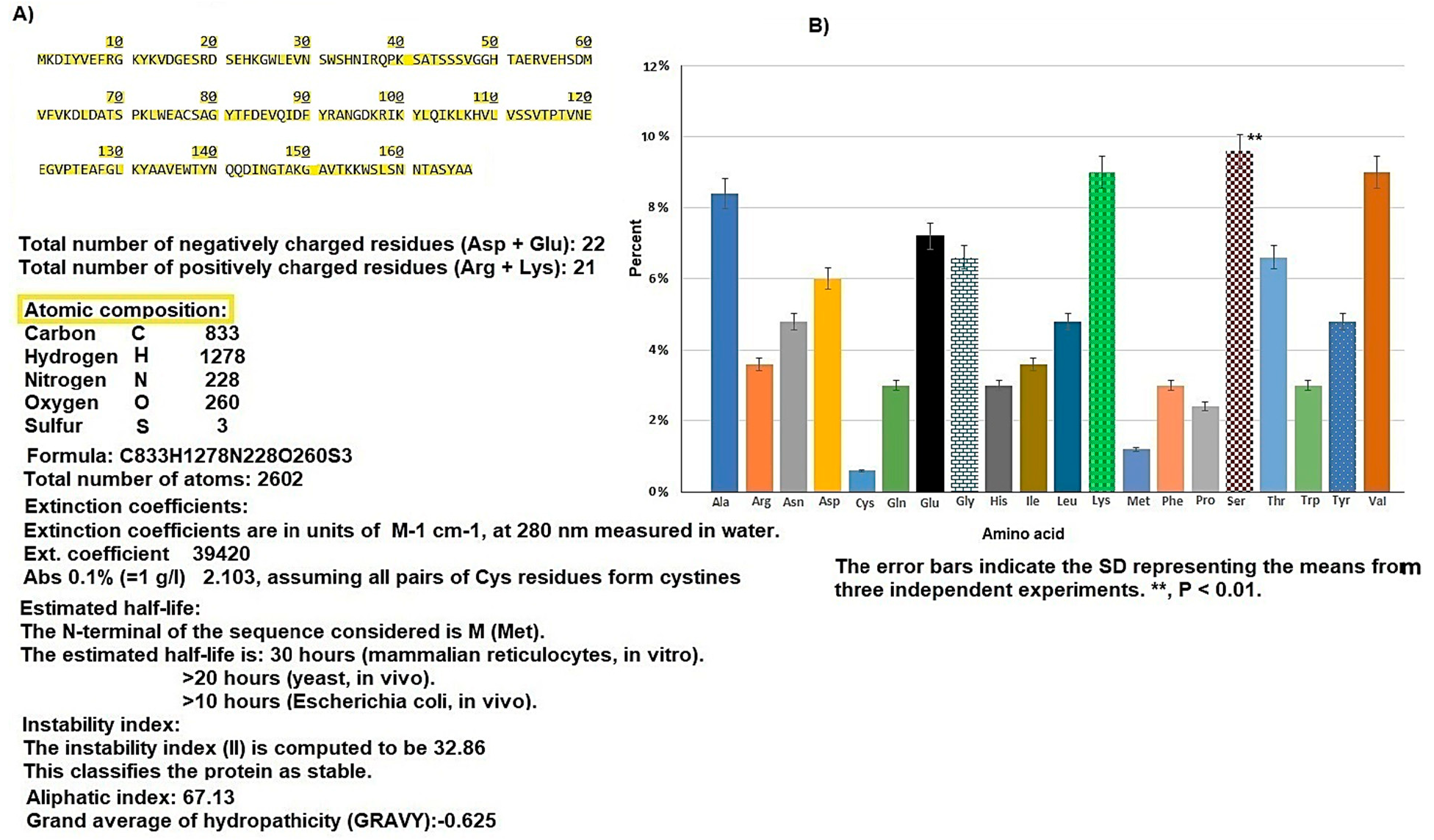
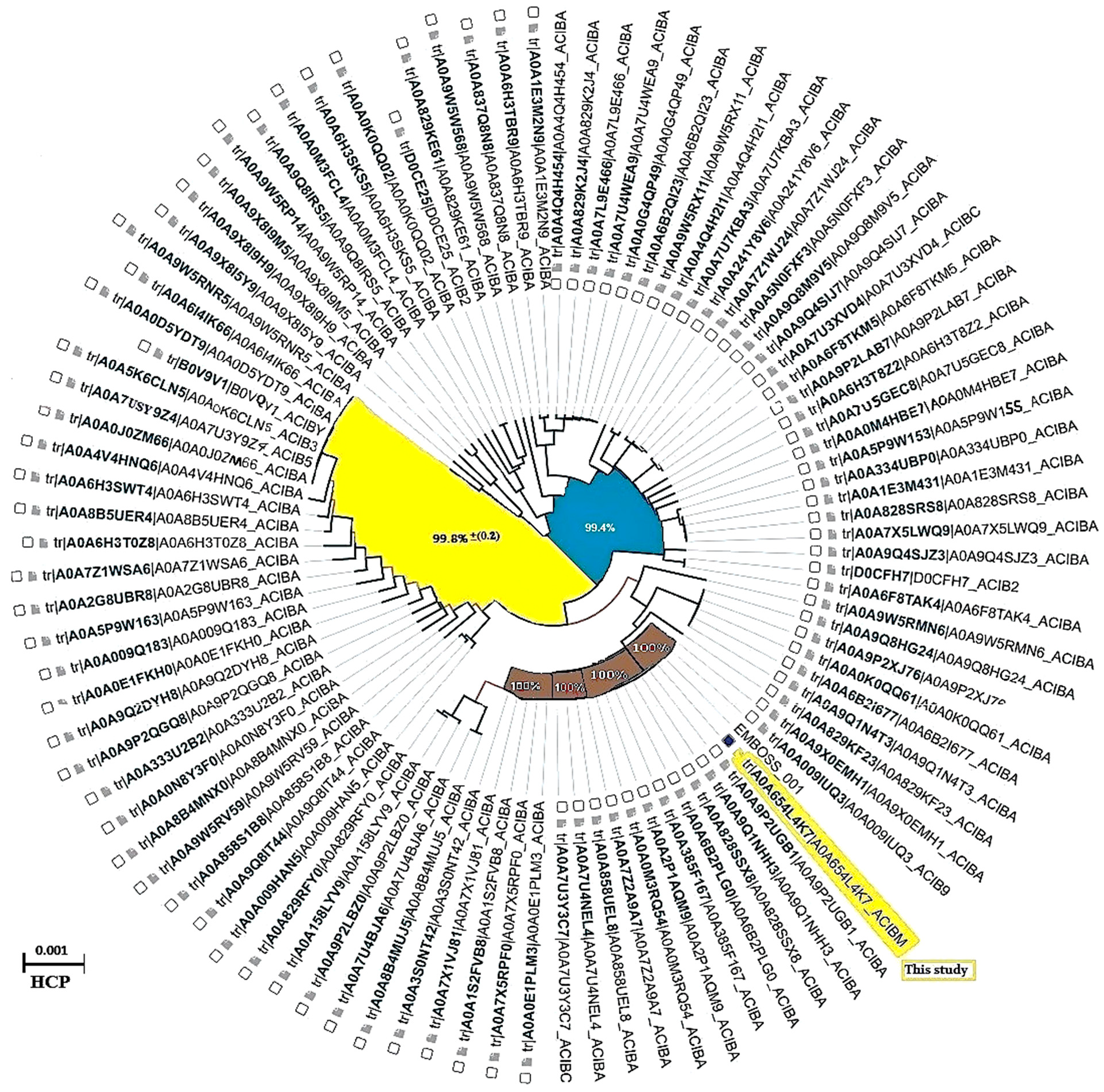
3.1. Hcp1 Homohexameric 3D Model Analysis The cartoon illustrations of homohexamer Hcp1 from A. baumannii (Figure 1A) indicated that the Hcp1 protein has multiple β-sheet barrels oriented in opposing directions, linked by short loops with the hexagonal ring. The structure generated using AlphaFold2 DeepMind revealed a tight β-barrel fold domain formed by two β sheets and one eight-residue α helix (pLDDT ≥ 90). Further analysis of the Hcp1 homohexamer (Figure 1B) by the line model highlighted the amino acids involved in binding to the TagX toxic effector molecule, arranged on the interior side of Hcp1 and situated within the conserved region of Hcp1. Indeed, the hydrophobic amino acids are buried within the Hcp1 ring, while hydrophilic amino acids are arranged in the exterior region (Figure 1B). The virtual analysis of the Hcp1 molecule also demonstrated that the inner diameter of the Hcp1 tail/tube complex in A. baumannii is estimated to be 40 Å, while the outer diameter measures around 80 Å, arranged within an asymmetric unit. These results were supported by control measurements that assessed the accuracy of the predicted model, which included Z-score, RMSD index, B-factor, melting point, and MolProbity in the Ramachandran plot. The template demonstrated a high-quality structure with a Z-score of 1.7. The predicted alignment error (PAE) map, often used in protein structure prediction, can be compared to distance variations (DV) obtained from molecular dynamics (MD) simulations. If the PAE map shows minimal DV, it suggests a high level of agreement between the predicted structure and the dynamic behavior observed in the MD simulations. This implies that the predicted structure is a good representation of the protein’s conformational landscape as determined by its PAE plot. In our case, it was a 5–7 scored residue (Figure 1C). At the same time, pairwise amino acid alignments of the Hcp1 protein show a conserved non-redundant region comprising a putative toxic effector domain with 100% identity compared to five closely related strains obtained from the UniProtKB database (Figure 1D). The color column and respective arrows indicate the position and kind of amino acids that bind to the effector molecule. Further analysis by the hidden Markov model (HMM) supports the above results by revealing all likelihood sequences for possible combinations of gaps, matches, and mismatches. 3.2. Analysis of the Secondary Structure of Hcp1 Homohexamer Meanwhile, to explore the number of chains of Hcp1 monomer, the schematic Spacefile structure of the Hcp1 homohexamer chains predicted in this study is demonstrated in Figure 2A. The presence of six chains designated as A, B, C, D, E, and F indicates that the assembly of the Hcp1 homohexamer in A. baumannii is a multi-complex process that necessitates additional components, such as the presence of an oligomerization domain in the T6ssK protein N-terminal. Chains E and F are located on the posterior side of the hexamer. The structure of Hcp1 was predicted by AlphaFold2 using PyMOL in the Schrödinger platform, showing that the extended loop connecting β2-sheet to β7 acts as a cap for the ring stabilization and interacts with the initial residues of Hcp1 (Figure 2B). Two loops, 1 and 2, in the middle of the model help with the stability of Hcp1 from the outer membrane to the exterior region. Further analysis indicated that the β6, β7, β8, and short α-helix play a role in Hcp1 chaperone activity, as shown by X-ray crystallography [19]. The important observation in this research was that the β3, β4, β7, and β8 sheets stack together to form a β-barrel with a hydrophobic core, as it was detected by the amino acid bonds to the effector. The quality and reliability of the present study model were assessed based on parameters such as pLDDT and PAE values as described in the methods. The results revealed that the first β-plate is formed by strands β1, β4, β6, and β8, are packed against the second β-sheet by strands β2, β3, β5, and β7, respectively, thus forming a β-barrel fold. To find the percent angstrom-level errors in the dihedral angles of the amino acid backbone and side chains involved in binding to the toxic effector, the RoseTTAFold program was employed as shown in Figure 2C. One peak error was found at position 50 in the graph, signifying the accuracy in amino acid residual conformation changes. 3.3. Characterization of the Toxic Effector TagX The elimination of target cells has been observed across various species of Acinetobacter, specifically A. nosocomialis M2, A. baumannii, and A. baylyi ADP1. In this study, the peptidoglycan L-alanyl-D-glutamate cell wall hydrolase, known as TagX, from A. baylyi ADP1 was selected as a template toxic effector (Figure 3). The amino acid sequence of the TagX enzyme was obtained from Weber et al. [18] and was employed to construct a three-dimensional structure of this toxic effector in this study. The structure of TagX effector consisted of one α-helix linked by several loops to four small β-sheets, which were located at the N-terminal of the enzyme and two domains, one connected to the α-helix in the form of a transmembrane nexus, and the other functioning as a peptidase activity. This peptidase domain is a bacterial peptidoglycan hydrolase that cleaves the peptide bonds within the peptidoglycan matrix, leading to cell lysis. It functions similarly to Tle1 from P. aeruginosa, another peptidoglycan hydrolase that targets the same structure and causes cell death [18]. 3.4. Molecular Docking and Conformational Changes in the Hcp1 A key aspect of this study was to determine how the toxic effector is transported through the injectosome spanning the inner to outer membrane (VgrG1 + PAAR + Hcp1) and the neighboring cell. To address this issue, the TagX effector molecule was docked to specific amino acids within the Hcp1 monomer using AutoDock Vina within the Schrödinger platform. The docking analysis revealed that four amino acids, Arginine, Serine, Histidine, and Glutamic acid, are playing a central role in binding to TagX α-helix and β-sheet, forming hydrogen bonds (Figure 4A). The estimated rotational angles of the bond amino acids showed a rotameric shift in the dihedral angles of these four amino acids His (18 Å), Arg 98 (9 & 8.6 Å), Ser 78 (13 Å), and Glu (12 Å) within the Hcp1 ring, respectively. Only in the case of Arg 98, we found the rotameric shift in the dihedral angle of both backbone and side chain. This might be due to the nature of the amino acid or effector compound, which affects the Cβ and N bond (φ) in the χ2 axis. These angles may also indirectly influence the side chains of amino acids, such as χ1 (between Cα and Cβ) and χ2 (between Cβ and Cγ). These rotameric shifts in dihedral angles of the above amino acids lead to changes in the conformation of amino acids within Hcp1 so that it activates other components of T6SS, like VgG29 contractile sheath and CLpV ATPase proteins, and causes contraction of 600 Hcp1 nanotubes. Ramachandran plot of Hcp1 monomer docked with TagX depicted in Figure 4B, whereas a Cartoon depiction of amino acids on β6 and β8 strands that form H-bonds with TagX effector molecule are illustrated in Figure 4C, respectively. 3.5. Prediction of the Entire Structure of Hcp1 by AI The entire A. baumannii schematic 3D structure of the Hcp1, as well as the N-terminal oligomerization domain of T6ssK protein, are depicted in Figure 5A, and 5B. As Figure 5A indicates, the complete structure of the Hcp1 protein consists of 13 homohexamers stacked on top of each other sequentially, forming a 600 nm Hcp1 nanotube through which the toxic effector TagX is transported to the injectosome in the tip of T6SS. The Hcp1, a transmembrane tail/tube protein, is connected to the baseplate via VgrG (gp27) contractile sheath to the outer membrane VgrG (gp5), and PAAR complexes. The T6SS TssB/TssC proteins are required for 600 nm tail/tube stabilization to form a stable injectosome. Meanwhile, the 3D structure of TssK shows it is a trimeric protein containing 3 domains, one situated in the N-terminal region of the protein is homologous to phage receptor binding site and helps the oligomerization of the Hcp1 monomers, a central α-helix region, and a transmembrane binding site (Figure 5B). 3.6. T6SS Prediction in A. baumannii by Artificial Intelligence The entire structure of the type VI secretion system in A. baumannii was generated using a machine learning AI system (Figure 6A,B). These figures show the latest in silico structure prediction of the T6SS report utilizing artificial intelligence. Here, the position of the individual component of the system was illustrated along with the complete T6SS structure. The inner membrane is composed of a baseplate containing 3-protein complexes (T6ssK, T6ssF, T6ssG). This complex is associated with the transmembrane proteins (T6ssL, T6ssM, T6ssJ) as an asymmetrical entity. Furthermore, TssB and TssC form a stable dimer that is the repeated unit for sheath polymerization (Figure 6B). Six TssB/C dimers wrap around the Hcp tail/tube and stabilize the 600 nm Hcp1 nanotube. Effectors delivered by the T6SS are loaded within the inner tube, transported to the needle-like spike complex, and then injected into targeted prokaryotic and/or eukaryotic cells, which are situated on the tip of the T6SS macromolecule. 3.7. Determination of Hcp1 Chemical Composition and Amino Acids Constitution The ProtParam software version 2.0.5 was used to determine several physicochemical properties of Hcp1, including its extinction coefficient, instability index, aliphatic index, and GRAVY value. The results indicated an extinction coefficient of 39,420 M−1cm−1, an instability index of 32.86, an aliphatic index of 67.13, and a GRAVY value of −0.625. These parameters provide insights into the protein’s stability, hydrophobicity, and overall characteristics (Figure 7A). Additionally, the percent frequency of individual amino acids in the Hcp1 structure was also estimated by the same software and presented in Figure 7B. The results indicate that serine, with a frequency of 9.6%, was the most abundant amino acid, while cysteine, with a frequency of 0.6%, was the least abundant amino acid detected in this study. Tyrosine, valine, and lysine were identified as the most frequent amino acids in Hcp1 protein, respectively. 3.8. Phylogenetic Tree Analysis The phylogenetic tree relationship among the Hcp1 type VI secretion system in A. baumannii strains was investigated, as shown in Figure 8. The dendrogram was compared with closely related strains of this bacterium in the UniProtKB database. The results indicated that the Hcp1 protein in A. baumannii is highly conserved, with an identity of ≥90% with those sequences reported in the UniProtKB database. It showed 100% identity with A. baumannii strains like A0A9P2UB1_ACIBA, A0A9Q1NHH3_ACIBA, and A9A9Q1NHH3_ACIBA, and 99.6% identity with 25 other Hcp1 sequences in this database (Figure 8).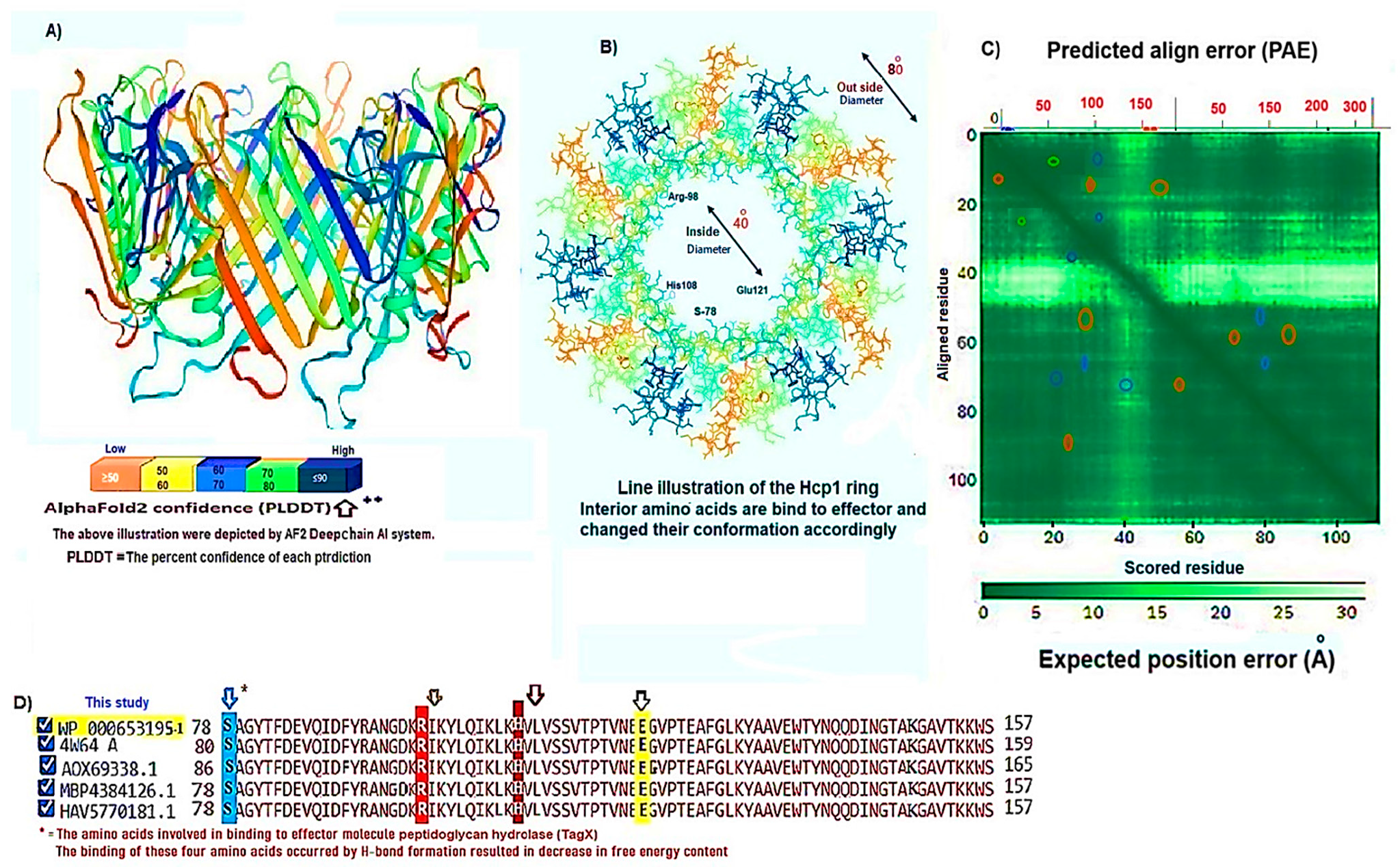
![Figure 3: <p>The schematic representation and amino acid composition, and residues of the primary domains of TagX peptidoglycan hydrolyase (pfam13539) are outlined. The structure feature contains an α-helix that functions as a transmembrane domain, which is linked to four β-sheet barrels via multiple loops. The β-sheets are responsible for the endopeptidase activity observed in this enzyme. Notably, the β3 sheet is located at the C-terminal end of the protein. The structural model of this toxin was developed using AlphaFold2, with <em>A. baylyi</em> ADP1 serving as the template organism. The model alignment demonstrated a pLDDT value of 90 or higher. The amino acid sequence for the TagX effector was sourced from the work of Weber et al. (2016) [<a href="#ref18">18</a>].</p>](/uploads/source/articles/molecular-modeling-connect/2025/volume2/20250009/image003.png)
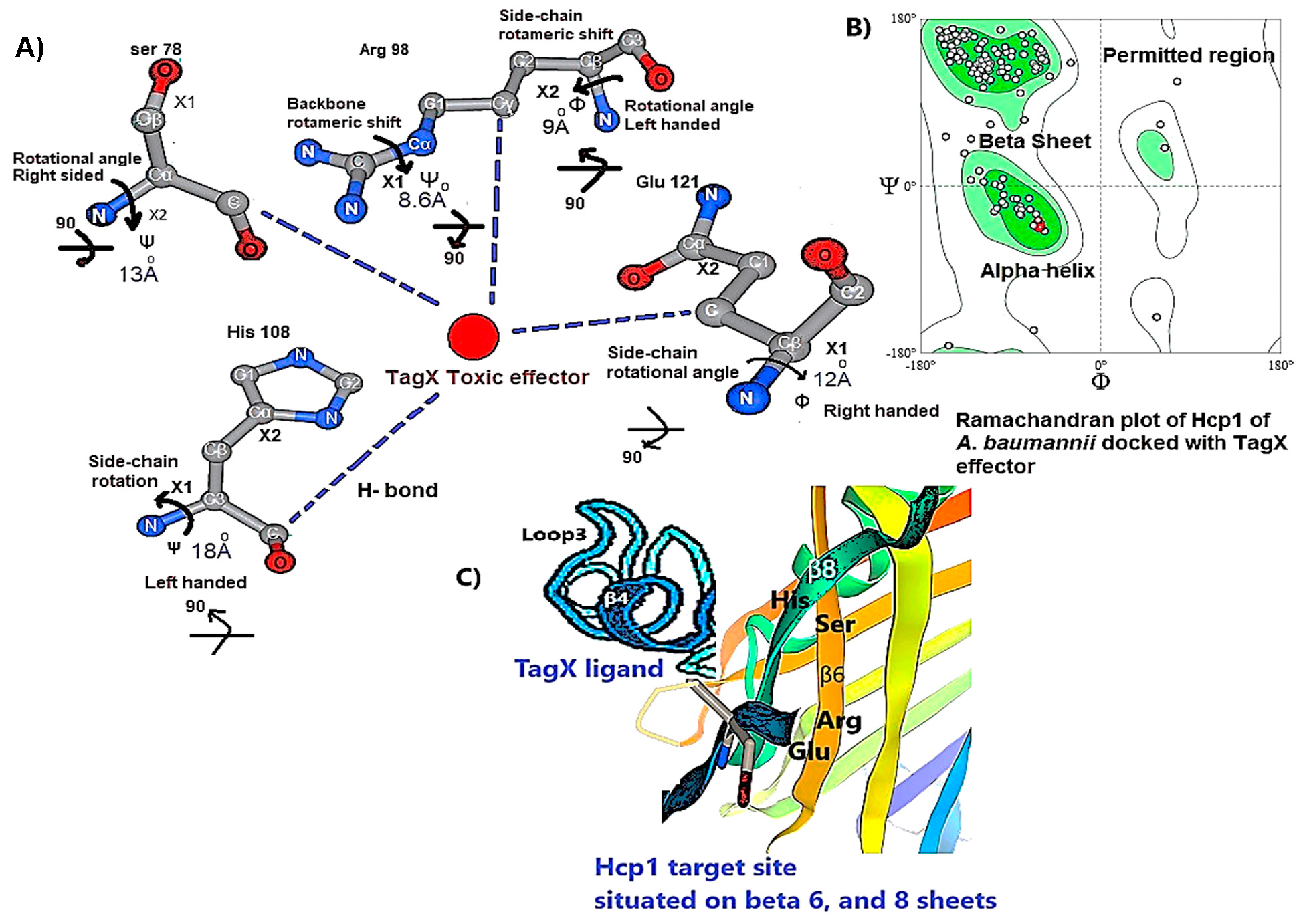
While different classes of the Type VI Secretion System have been identified in A. baumannii, they all share a fundamental structure and function [19]. The Hcp1 is described to maintain notable structural and functional similarities to the tail of the T4 bacteriophage, indicating a potential common evolutionary lineage. This relationship not only enhances our understanding of the evolutionary dynamics underlying these biological systems but also suggests possible implications for their roles in microbial interactions and pathogenicity [20]. The structure of Hcp1 protein exhibits two β sheets that run to each other in reverse orientation and are flanked at one side by a short α-helix. Six Hcp molecules associate to form a donut-shaped hexamer, as observed in both the crystal structure and solution in a symmetric orientation. Recognizing these parallels could provide valuable insights into the adaptive mechanisms employed by microorganisms, thereby affirming their significance as a critical virulence factor [10]. The majority of T6SS-producing bacteria exist in soil where there is a scarcity of the presence of micronutrients among bacterial populations. The evidence suggests that some bacteria have evolved mechanisms for predation and competition which were later adapted for use as virulence factors in pathogens. These mechanisms, initially developed for survival in diverse environments, can be detrimental to other organisms, including potential hosts, when employed during infection. This phenomenon, known as coincidental evolution, implies that virulence factors may not have evolved specifically for pathogenicity, but rather for other ecological roles that subsequently became useful in a parasitic lifestyle [21]. Therefore, the presence of the T6SS is of utmost importance for the survival of host cells by killing neighboring cells. The Hcp protein is both a core component of the T6SS tail tube and the exported receptor/chaperone of the effectors [22]. However, there is little information regarding the mechanism triggering contraction of the Hcp1 nanotube complex protein and how the toxic effector-Hcp1 complex was propelled into the targeted cell [23]. In the present study, the changes in the dihedral angles of both the side-chains and backbones of the amino acids Lys, Phe, Arg, and His within the Hcp1 molecule, resulting from the entry of TagX effector into the Hcp1 nanotube, were identified. Backbone rotation is primarily due to steric repulsion between backbone atom Cγ, whose position depends on φ and ψ side-chain heavy atoms. The conformational changes were identified using the RoseTTAFold AI system, which typically reports the specific amino acid, corresponding dihedral angles, measures of variance, and the likelihood of occurrence [24]. Further analysis found that TagX from A. baylyi is an efficient inducer of the rotameric shift in the amino acids side-chains within Hcp1 monomer and creating torsional forces and H-bound formation between the ligand molecule and receptors. This process ultimately stimulates the contraction of the Hcp1 tail/tube complex, aided by VgrG1(gp27) contractile sheath and ATP hydrolysis, thereby propelling the effector molecule into the target cell. The internal pore, part of the Hcp hexamer, is capable of accommodating small, folded proteins (under 20 kDa) or unfolded proteins. This includes proteins like Tse1-3 from P. aeruginosa and EvpC from Edwardsiella tarda, which are known to bind to the inner surface of the Hcp hexamer. Specifically, the Hcp hexamer, with its β-barrel domain, forms a ring-like structure with an inner diameter of approximately 40 Angstroms. This size limitation allows for the transport of smaller proteins through the pore, while larger, folded proteins are excluded [25]. The significant contribution of this study lies in creating a three-dimensional model of the TagX effector molecule. This model is based on the amino acid sequence data reported by Weber et al. [18]. Their work revealed that the TagX effector molecule contains two main domains: a peptidoglycan hydrolase domain, which hydrolyzes the cell wall and causes lysis, and another domain containing a transmembrane helix connected through β-sheets to long loops. A recent study indicates that in Xanthomonas bovienii, Hcp1 but not Hcp2 is required to suppress the prey bacterial populations. In one case, it appears that the mutation Arg-to-Ala at position 30 within the extended Hcp1 resulted in a significant decrease in cytotoxicity, suggesting that mutations of Hcp1 shut down the functioning of all T6SS components [26,27]. Overall, the application of AI in epidemiology, microbial ecology, and forensic microbiology is also outlined, emphasizing its proficiency in deciphering complex microbial interactions and forecasting disease outbreaks. We critically examined the challenges in AI applications, such as ensuring data quality and overcoming algorithmic constraints, and stressed the necessity for interpretable AI models that align with medical and ethical standards [28].
The study focuses on how TagX interacts with Hcp1 nanotubes, which are crucial for the delivery of toxic molecules by the T6SS (translocation system). The interaction leads to Hcp1 nanotubes contracting and propelling the effector molecule toward the tip of the injectosome (the T6SS). AlphaFold2 and bioinformatics software were used to elucidate the mechanism, revealing that TagX binding induces rotameric shifts and conformational changes in Hcp1, ultimately triggering contraction of 600 nanotubes via the VgrG1 sheath, TssM, and ATP hydrolysis. This research also highlights the potential of machine learning in drug discovery by identifying a promising candidate for targeting Gram-negative pathogens.
| A2 | AlphaFold2 |
| A. baumannii | Acinetobacter Baumannii |
| Hcp | Hemolysin Coregulator Protein |
| PAAR | Proline-Alanine-Alanine-Arginine |
| PAE | Predicted Aligned Error |
| T6SS | Type 6 Secretion System |
M.R.S.: Conceptualization, Data curation, Methodology. S.S.: Writing original draft, contributing to data mining and software analysis. All authors have read and agreed to the published version of the manuscript.
The raw data supporting the conclusions of this article is made available by the authors, without undue reservation, in preprint at the Research Square site https://www.researchsquare.com. Posted Date: October 23rd, 2024 DOI: https://doi.org/10.21203/rs.3.rs-5231960/v1.
No consent for publication is required, as the manuscript does not involve any individual personal data, images, videos, or other materials that would necessitate consent.
The authors declare that no conflicts of interest exist.
No fund was used for this research.
The authors would like to thank the staff of Research Center for Infectious Diseases and Tropical Medicine, Kerman University of Medical Sciences, Iran, Medical Mycology and Bacteriology Research Center and Department of Microbiology and Virology, Kerman University of Medical Sciences for their help and computer assist in this investigation.
[1] Le, N.H.; Pinedo, V.; Lopez, J.; Cava, F.; Feldman, M.F. Killing of Gram-negative and Gram-positive bacteria by a bifunctional cell wall-targeting T6SS effector. Proc. Natl. Acad. Sci. USA 2021, 118, e2106555118. [CrossRef] [PubMed]
[2] Jiang, X.; Li, H.; Ma, J.; Li, H.; Ma, X.; Tang, Y.; Li, J.; Chi, X.; Deng, Y.; Zeng, S.; et al. Role of Type VI secretion system in pathogenic remodeling of host gut microbiota during Aeromonas veronii infection. ISME J. 2024, 18, wrae053. [CrossRef] [PubMed]
[3] Yang, X.; Long, M.; Shen, X. Effector Immunity Pairs Provide the T6SS Nanomachine its Offensive and Defensive Capabilities. Molecules 2018, 23. [CrossRef]
[4] Carruthers, M.D.; Nicholson, P.A.; Tracy, E.N.; Munson, R.S. Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS ONE 2013, 8. [CrossRef]
[5] Motta, E.V.S.; Lariviere, P.J.; Jones, K.R.; Song, Y.; Moran, N.A. Type VI secretion systems promote intraspecific competition and host interactions in a bee gut symbiont. Proc. Natl. Acad. Sci. USA 2024, 121, e2414882121. [CrossRef]
[6] Sharma, S.; Raju, S.; Verma, S.K.; Kamal Verma, R.; Thakur, P.K.; Sharath Kumar, K.S. Pyrazoles: A Master Key to Tackle Multidrug-Resistant Acinetobacter baumannii and Its Structure Activity Relationship Studies. Chem. Biol. Drug Des. 2025, 105. [CrossRef] [PubMed]
[7] Wang, T.; Hu, Z.; Du, X.; Shi, Y.; Dang, J.; Lee, M.; Hesek, D.; Mobashery, S.; Wu, M.; Liang, H. A type VI secretion system delivers a cell wall amidase to target bacterial competitors. Mol. Microbiol. 2020, 114, 308–321. [CrossRef]
[8] Peng, Y.; Wang, X.; Shou, J.; Zong, B.; Zhang, Y.; Tan, J.; Chen, J.; Hu, L.; Zhu, Y.; Chen, H.; et al. Roles of Hcp family proteins in the pathogenesis of the porcine extraintestinal pathogenic Escherichia coli type VI secretion system. Sci. Rep. 2016, 6. [CrossRef]
[9] Ruiz, F.M.; Santillana, E.; Spínola-Amilibia, M.; Torreira, E.; Culebras, E.; Romero, A. Crystal structure of Hcp from Acinetobacter baumannii: A component of the Type VI secretion system. PLoS ONE 2015, 10. [CrossRef]
[10] Ruiz, F.M.; Lopez, J.; Ferrara, C.G.; Santillana, E.; Espinosa, Y.R.; Feldman, M.F.; Romero, A. Structural Characterization of TssL from Acinetobacter baumannii: A Key Component of the Type VI Secretion System. J. Bacteriol. 2020, 202, e00210-20. [CrossRef]
[11] Silverman, J.M.; Brunet, Y.R.; Cascales, E.; Mougous, J.D. Structure and regulation of the type VI secretion system. Annu. Rev. Microbiol. 2012, 66, 453–472. [CrossRef] [PubMed]
[12] Ma, J.; Pan, Z.; Huang, J.; Sun, M.; Lu, C.; Yao, H. The Hcp proteins fused with diverse extended-toxin domains represent a novel pattern of antibacterial effectors in type VI secretion systems. Virulence 2017, 8, 1189–1202. [CrossRef]
[13] Lin, Y.; Zhao, D.; Huang, N.; Liu, S.; Zheng, J.; Cao, J.; Zeng, W.; Zheng, X.; Wang, L.; Zhou, T.; et al. Clinical impact of the type VI secretion system on clinical characteristics, virulence and prognosis of Acinetobacter baumannii during bloodstream infection. Microb. Pathog. 2023, 182, 106252. [CrossRef] [PubMed]
[14] Kim, J.; Lee, J.Y.; Lee, H.; Choi, J.Y.; Kim, D.H.; Wi, Y.M.; Peck, K.R.; Ko, K.S. Microbiological features and clinical impact of the type VI secretion system (T6SS) in Acinetobacter baumannii isolates causing bacteremia. Virulence 2017, 8, 1378–1389. [CrossRef]
[15] Ma, J.; Sun, M.; Pan, Z.; Song, W.; Lu, C.; Yao, H. Three Hcp homologs with divergent extended loop regions exhibit different functions in avian pathogenic Escherichia coli. Emerg. Microbes Infect. 2018, 7, 49. [CrossRef] [PubMed]
[16] Li, M.; Le Trong, I.; Carl, M.A.; Larson, E.T.; Chou, S.; De Leon, J.A.; Dove, S.L.; Stenkamp, R.E.; Mougous, J.D. Structural basis for type VI secretion effector recognition by a cognate immunity protein. PLoS Pathog. 2012, 8. [CrossRef]
[17] Chou, S.; Bui, N.K.; Russell, A.B.; Lexa, K.W.; Gardiner, T.E.; LeRoux, M.; Vollmer, W.; Mougous, J.D. Structure of a peptidoglycan amidase effector targeted to Gram-negative bacteria by the type VI secretion system. Cell Rep. 2012, 1, 656–664. [CrossRef]
[18] Weber, B.S.; Hennon, S.W.; Wright, M.S.; Scott, N.E.; de Berardinis, V.; Foster, L.J.; Ayala, J.A.; Adams, M.D.; Feldman, M.F. Genetic Dissection of the Type VI Secretion System in Acinetobacter and Identification of a Novel Peptidoglycan Hydrolase, TagX, Required for Its Biogenesis. mBio 2016, 7, e01253-16. [CrossRef]
[19] Li, P.; Zhang, S.; Wang, J.; Al-Shamiri, M.M.; Han, B.; Chen, Y.; Han, S.; Han, L. Uncovering the Secretion Systems of Acinetobacter baumannii: Structures and Functions in Pathogenicity and Antibiotic Resistance. Antibiotics 2023, 12. [CrossRef]
[20] Veesler, D.; Cambillau, C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [CrossRef]
[21] Basler, M.; Mekalanos, J.J. Type 6 secretion dynamics within and between bacterial cells. Science 2012, 337, 815. [CrossRef]
[22] Mougous, J.D.; Cuff, M.E.; Raunser, S.; Shen, A.; Zhou, M.; Gifford, C.A.; Goodman, A.L.; Joachimiak, G.; Ordoñez, C.L.; Lory, S.; et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 2006, 312, 1526–1530. [CrossRef] [PubMed]
[23] Allsopp, L.P.; Bernal, P. Killing in the name of: T6SS structure and effector diversity. Microbiology 2023, 169. [CrossRef] [PubMed]
[24] Meng, Y.; Zhang, Z.; Zhou, C.; Tang, X.; Hu, X.; Tian, G.; Yang, J.; Yao, Y. Protein structure prediction via deep learning: An in-depth review. Front. Pharmacol. 2025, 16. [CrossRef] [PubMed]
[25] Zheng, J.; Leung, K.Y. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 2007, 66, 1192–1206. [CrossRef]
[26] Song, C.; Yang, B. Mutagenesis of 18 type III effectors reveals virulence function of XopZ(PXO99) in Xanthomonas oryzae pv. oryzae. Mol. Plant Microbe Interact. 2010, 23, 893–902. [CrossRef]
[27] Raza, M.A.; Farwa, U.; Danish, M.; Ozturk, S.; Aagar, A.A.; Dege, N.; Rehman, S.U.; Al-Sehemi, A.G. Computational modeling of imines based anti-oxidant and anti-esterases compounds: Synthesis, single crystal and In-vitro assessment. Comput. Biol. Chem. 2023, 104. [CrossRef]
[28] Tsitou, V.M.; Rallis, D.; Tsekova, M.; Yanev, N. Microbiology in the era of artificial intelligence: Transforming medical and pharmaceutical microbiology. Biotechnol. Biotechnol. Equip. 2024, 38. [CrossRef]