
 ACCESS
Review Article
ACCESS
Review Article
Volume 1, Article ID: 2025.0009

Walaa K. Mousa
walaa.mousa@aau.ac.ae


Aya Al Ali
202221536@aau.ac.ae


Ranim W. A. Abdelmoteleb
ranim.abdelmoteleb@aau.ac.ae


Ruqaia Al Shami
ruqaia.alshami@aau.ac.ae


Najwa Al Ramadan
201920320@aau.ac.ae


Sedq Moutraji
sedq.moutraji@aau.ac.ae

Rose Ghemrawi
rose.ghemrawi@aau.ac.ae
1 College of Pharmacy, Al Ain University, Abu Dhabi 64141, UAE
2 AAU Health and Biomedical Research Center, Al Ain University, Abu Dhabi 112612, UAE
3 College of Pharmacy, Mansoura University, Mansoura 35516, Egypt
* Author to whom correspondence should be addressed
Received: 27 Apr 2025 Accepted: 26 May 2025 Available Online: 26 May 2025 Published: 17 Jun 2025
The human body harbors distinct microbial communities at each body site. One microbial niche of particular interest within the human body is the tumor microenvironment. These intratumor microbes are linked to tumor initiation, progression, and metastasis through diverse mechanisms, including activation of oncogenic pathways and modulation of antitumor immunity. Recent studies have emphasized the role of intratumor microbes in influencing the response and outcome of cancer immunotherapeutics and vaccines. Further data suggest a crucial role of microbial metabolites in the metabolic rewiring of CD8+ T cells controlling antitumor immunity. This knowledge is vital to promote our understanding of the role of microbes in the tumor microenvironment and advance translational applications. In this review, we discuss factors that shape the structure of the intratumor microbiome, such as the translocation of gut microbes and the development of local microbial communities. This study highlights the remote control of gut microbes in the tumor microenvironment, disease progression, and therapy outcome. We detail interactions of intratumor microbes and their crosstalk with tumor and immune cells, such as tissue-resident and tumor-infiltrating T cells. We discuss open research questions in this field, including defining oncomicrobiotics, the subset of microbiota with biotherapeutic potential in inducing antitumor immunity. We highlight challenges and opportunities, emphasizing the future direction of developing next-generation engineered probiotics that can advance delivery, maximize the efficacy of cancer therapy, or even serve as a non-invasive technique to sense and detect tumor cells.
Tumor microenvironment (TME) plays a major role in shaping tumor behavior. Microbes residing inside the tumor cells, referred to as intratumor microbiome or oncobiomes, play a crucial role in tumor development, progression, response to therapeutics, prognosis, and clinical outcome [1,2,3,4,5]. Recent research has shown that these microbes may either translocate from the gut or oral cavity following dysbiosis or be local residents that thrive in the tumor microenvironment due to its immunosuppressive nature and the presence of a leaky vascular network within cancerous lesions [6,7]. Intratumor microbes can manipulate the anti-tumor immunity in multiple types of cancers, including colon, pancreas, prostate, breast, and lung [1,2,3,4,5] (Figure 1). Intratumor microbes can reprogram cytotoxic CD8+ T cells, among other immune cells, and affect their tumor infiltration rates. In addition, these microbes control the levels of proinflammatory cytokines. Multiple studies suggest that tumor-infiltrating tissue-resident memory T cells (TRM) are crucial to achieve the desired response in solid tumors [8,9,10,11] and especially in patients receiving PD-1 therapy [12]. Interestingly, memory responses by IFN–γ–secreting CD8+ and CD4+ T cells specific for Bacteroides fragilis, Enterococcus hirae, and Akkermansia muciniphila correlated with positive outcomes in cancer therapy [13,14,15,16]. Microbiome signatures, whether in the gut or within the tumor microenvironment (TME), are gaining attention as a major factor contributing to interpatient heterogeneity and influencing the response to immunotherapies such as anti-PD-1. For example, the abundance of Collinsella aerofaciens, Bifidobacterium longum, and E. faecium in the feces of patients is linked to a good response to anti-PD-1, and their fecal transplant to germ-free (GF) mice resulted in increased T cells and improved therapy outcome [4]. In this review, we showcase the recent advances in understanding the structure and function of the intratumor microbiome and its influence on the disease’s progression and therapy outcome. This study highlights the potential application of this knowledge to enhance the efficacy of immunotherapeutics or to develop novel microbiome-based diagnostic biomarkers and therapeutics.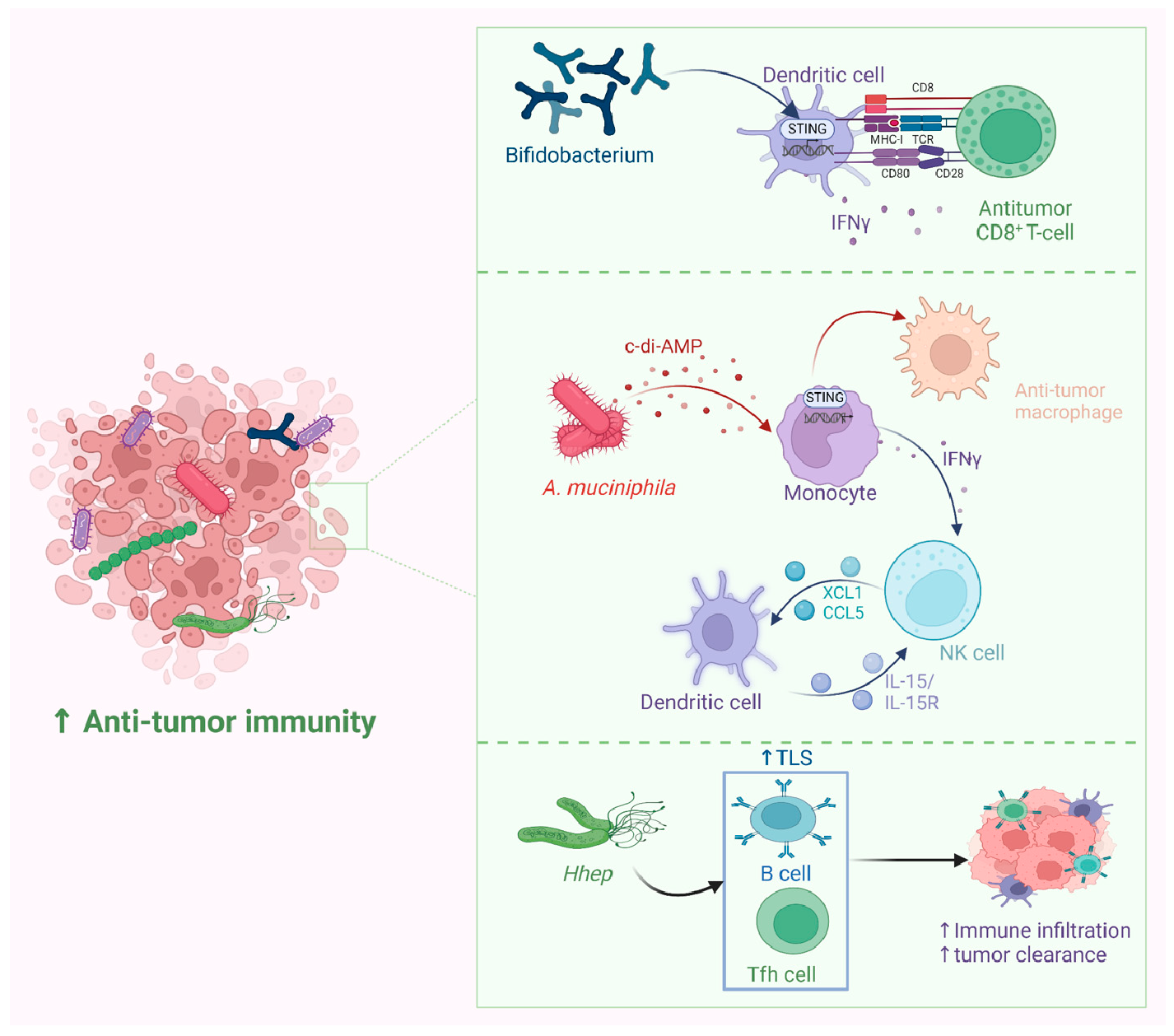
Various pathways facilitate microbiota access to the TME. Tumors originating in organs directly exposed to the external environment, such as in nasopharyngeal cancer, may carry bacteria from the local microbiome. In a cohort of 800 patients with nasopharyngeal cancer, higher intratumoral bacterial loads were associated with reduced survival rates. The analysis pinpointed the nasopharyngeal microbiota as the principal origin of intratumoral bacteria [17]. The disturbed epithelial or mucosal barrier in some tumors can promote the colonization of resident microbiota. For example, tumors with TP53 mutations, known to impede epithelial function, exhibit a distinct bacterial consortium, primarily featuring Acidovorax temporans in lung cancer [18]. Several factors enable the intracellular colonization of bacteria within the tumor cells. The hypoxic tumor microenvironment favors the survival of anaerobic and facultative anaerobic bacteria, with varying oxygen levels in different tissues contributing to differences in the residing bacteria [19]. Except for lung cancer, most cancers show a predominance of anaerobic bacteria. TME is an immunosuppressive region [20], which impairs immune-mediated clearance of bacteria. The disrupted vascular system is also a favorable condition for rapid bacterial entry and colonization in tumors [21]. Alongside this, tumor tissues with necrotic regions provide nutrients and molecules that support bacterial outgrowth [22]. A recent study showed that the diversity of the microbiome is linked to the biopsy site, emphasizing the influence of the surrounding environment, rather than the primary tumor type [23]. Further, bacteria possess the capability to disseminate from remote anatomical sites and establish colonization within tumor tissues via the bloodstream or other physical channels. For example, the breakdown of barriers caused by genetic lesions initiating colorectal cancer leads to the invasion of adenomas by microbiota and microbial products. These products, in turn, activate inflammation initiated by the tumor, fostering tumor growth [24]. Oral-originated microbiota, including four Fusobacterium spp., were found to be enriched in colorectal tumors [25].
Growing evidence supports that gut microbes translocate to the tumor site, where they reside and shape the tumor microbiome landscape. These microbes, together with local microbial residents, can rewire the CD8+ T cells and guide them to promote or inhibit anti-tumor immunity and hence determine tumor growth and outcome, together with the host factors and tumor genetics [26,27,28,29,30,31]. Diversity of gut microbiota is linked to local and distant immune signatures that could be either favorable or unfavorable in tumor progression and metastasis [32]. Dysbiosis and leaky gut create a chronic inflammatory status conducive to tumor development and progression [3]. Gut microbes play a crucial role in the maturation of the immune system and controling the anti-tumor immunity. During early gut colonization, commensal microbial antigens are transported to the thymus by dendritic cells, promoting T cell expansion [33]. More insights have been gained from studies on germ-free mice that support the notion that microbial antigens control the development of T cells [34]. Early evidence of the microbiota’s role in anti-tumor immunity emerged in 2007 with the discovery that commensals activate antigen-presenting cells via Toll-like receptor 4 (TLR4) [13]. Further, a study shows that TLR4 agonists modulate tumor necrosis factor (TNF) and initiate anti-tumor activity [14]. In addition, secreted microbial metabolites from the leaky gut can exert a remote control over the TME as detailed in the mechanisms below.
4.1. Microbiota Mediate a State of Chronic Inflammation Leading to Tumor Initiation Several studies suggest that dysbiosis in the gut contributes to oncogenesis, especially for cancers of the colon, liver, and pancreas. This effect is mediated by leaked microbial metabolites that modulate the host immune response [27,35,36]. A leaky gut and increased circulating levels of lipopolysaccharide (LPS) that create a chronic state of inflammation are drivers for cancer development. For example, increased levels of circulating microbial-derived LPS in obesity and type 2 diabetes are associated with a higher risk of colorectal cancer [37]. Another line of evidence shows that elevated levels of secondary bile acids in mice are linked to overexpression of cyclooxygenase-2 (COX-2), an enzyme involved in prostaglandin pathways, which are linked to inflammation and cancer [38]. Other metabolites derived from Clostridium spp. and implicated in inflammation pathways are lithocholic acid and muricholic acid, which suppress chemokine (C-X-C motif) ligand 16 (CXCL16) in the liver, hindering the recruitment of natural killer T (NKT) cells, resulting in tumor progression and metastasis in mice [35]. Interestingly, the administration of oral antibiotics that deplete Clostridium increased the expression of CXCL16, resulting in the accumulation of NKT cells and achieving more control over tumor growth [35]. The chronic inflammatory response worsens tissue damage and consequent influx of infiltrating microbes/microbial metabolites, resulting in excessive production of cytokines and chemokines, which might foster angiogenesis [39] (Figure 2). High levels of pro-inflammatory mediators were associated with the tumor microbiome [40]. A study on a genetically engineered mouse model found that the microbiota can induce inflammation and advance the progression of cancer by acting through the lung-resident γδ T cells. In this model, lung tumor growth was associated with an increase in total bacterial load and a decrease in bacterial diversity within the airway. Commensal bacteria increase Myd88-dependent IL-1β and IL-23 production from myeloid cells, stimulating the activation of Vγ6+Vδ1+ γδ T cells that produce IL-17 and other effector molecules, promoting inflammation and tumor cell proliferation. Moreover, neutralization of IL-17, a key effector molecule produced by γδ T cells, resulted in reduced neutrophil infiltration and tumor burden [41]. Another study reported that intratumor bacteria induced the production of IL-17, which promoted an influx of B cells and the development of tumors [42]. In an attempt to study the link between microbiome, inflammation, and cancer, Hoste et al. employed a mouse model of wound-induced skin cancer and studied the mechanism by which the skin microbiota contributes to inflammation and tumorigenesis. In the presence of skin microbes, the removal of various innate immune sensors, including TLR-5, TNF receptor (TNFR)-1/-2, and MYD-88, protects against tumorigenesis, with inflammation showing a correlation with tumor incidence. Notably, the administration of antibiotics hinders tumor formation, while flagellin injection induces tumors, both in a TLR-5 dependent manner [43]. 4.2. Microbes Modulate Host Immunity Affecting Tumor Progression Microbes can enhance the progression of tumors by modulating the activity of several pathways related to host immunity. One example is by altering the immune response towards the cancer cells. This leads to remodeling of the TME, inducing an immunosuppressive environment which makes the cancer cell unrecognizable or non-responding to the immune system. For example, intratumor F. nucleatum promotes tumor growth by mediating antitumor immunity, represented by suppression of tumor-infiltrating CD8+ T cells [44]. Mathiasen et al. showed that cytolethal distending toxin (CDT), produced by many pathogenic gram-negative bacterial species, can induce premature senescence in activated CD4 T cells [45]. This suggests that bacterial toxins reduce the anticancer response and promote the proliferation of cancer. Another mechanism for the modulation of the immune response is by activating TLRs by the bacterial antigens. Activation of TLR can promote the activation of certain proliferation and angiogenesis responses, such as STAT3, NFkB, and ROS [46]. Other microbial metabolites that contribute to anti-tumor immunity are short-chain fatty acids (SCFAs) via their direct interaction with CD8+ T cells, which leads to improving their capacity to differentiate and exert anti-tumor activity [47,48,49]. A study found that the SCFA-producing Ruminococcaceae family is associated with an increase in T cell accumulation inside the tumor [50]. In support of this finding, another study showed that fecal transplantation from metformin-fed mice (that showed a higher abundance of Ruminococcaceae) resulted in an elevated level of SCFAs coupled with a suppression of tumor proliferation in a murine model [51]. Another study identified a positive correlation between the abundance of SCFAs-producing Lachnoclostridium genus (originally resides in the gut), inside the tumor, and the concentration of intratumor cytotoxic CD8+ T cells mediated by overexpression of chemokines C-C motif chemokine ligand 5 (CCL5), CXCL9, and CXCL10 [52]. 4.3. Microbial Metabolites Can Induce DNA Damage and Initiate Cancer Development Bacteria produce metabolites, proteins, and molecules that aid in directly damaging and altering the stability of the host genome, thus contributing to the development of mutations. For example, colibactin is a metabolite produced by pks+ Escherichia coli; this metabolite acts as a DNA alkylator and causes double-strand breaks as a consequence of DNA crosslinks [53,54]. Colibactin possesses a unique mutational pattern in organoids treated with genotoxic pks+ E. coli, similar to the mutation present in 5876 human cancer genomes [55]. Bacteroides fragilis could promote DNA damage by secreting B. fragilis toxin (BFT), although without a distinct mutational profile [56,57]. Through cell culture and animal models, Goodwin et al. reported that BFT induces the expression of spermine oxidase (SMO), which is a polyamine catabolic enzyme, resulting in higher reactive oxygen species (ROS) and DNA damage. Furthermore, they showed that inhibition of elevated SMO in B. fragilis-infected mice significantly reduces chronic intestinal inflammation and inhibits colon tumorigenesis [58]. Recently, a new genotoxic small molecule secreted by colorectal cancer-associated species, Morganella morganii, was discovered by Cao et al., named indolamines. These metabolites elicit DNA damage in intestinal epithelial cells (IECs) and are implicated in the development of colon tumors in gnotobiotic mouse models [59]. The microbiome can also disrupt the body’s DNA damage response. During DNA replication, base-base mismatches and insertion-deletion loops can occur when the primer slips against the template strand during the synthesis of a new strand [60]. DNA mismatch repair (MMR) functions to correct these errors. The inactivation of DNA MMR, both genetically and epigenetically, has the potential to induce mutations in genes associated with cancer and subsequently contribute to the development of cancer [61]. MMR genes are found to be downregulated in response to Helicobacter pylori [62]. Interestingly, H. pylori infection induces expression of microRNAs (miRs), such as miR-150-5p, miR-155-5p, and miR-3163, which in turn modulate and target MMR genes, such as POLD3, MSH2, and MSH3, respectively [62]. 4.4. Microbes Control Signaling Pathways Involved in Carcinogenesis Several microbes secrete molecules that interact with host pathways involved in carcinogenesis. For example, H. pylori produces a protein called CagA, which modulates β-catenin to drive gastric cancer and prostate cancer. CagA-mediated β-catenin activation leads to up-regulation of genes involved in cellular proliferation, survival, migration, and angiogenesis [63,64,65]. F. nucleatum is a member of the oral microbiota and is associated with human cancers [66]. F. nucleatum expresses FadA, a bacterial cell surface adhesion component that binds host E-cadherin, leading to β-catenin activation [66]. Enterotoxigenic B. fragilis, which is enriched in some human colorectal cancers, can stimulate E-cadherin cleavage via Bft, leading to β-catenin activation [67]. Salmonella typhi strains secrete AvrA, which can activate epithelial β-catenin signaling and are associated with colonic cancers [68].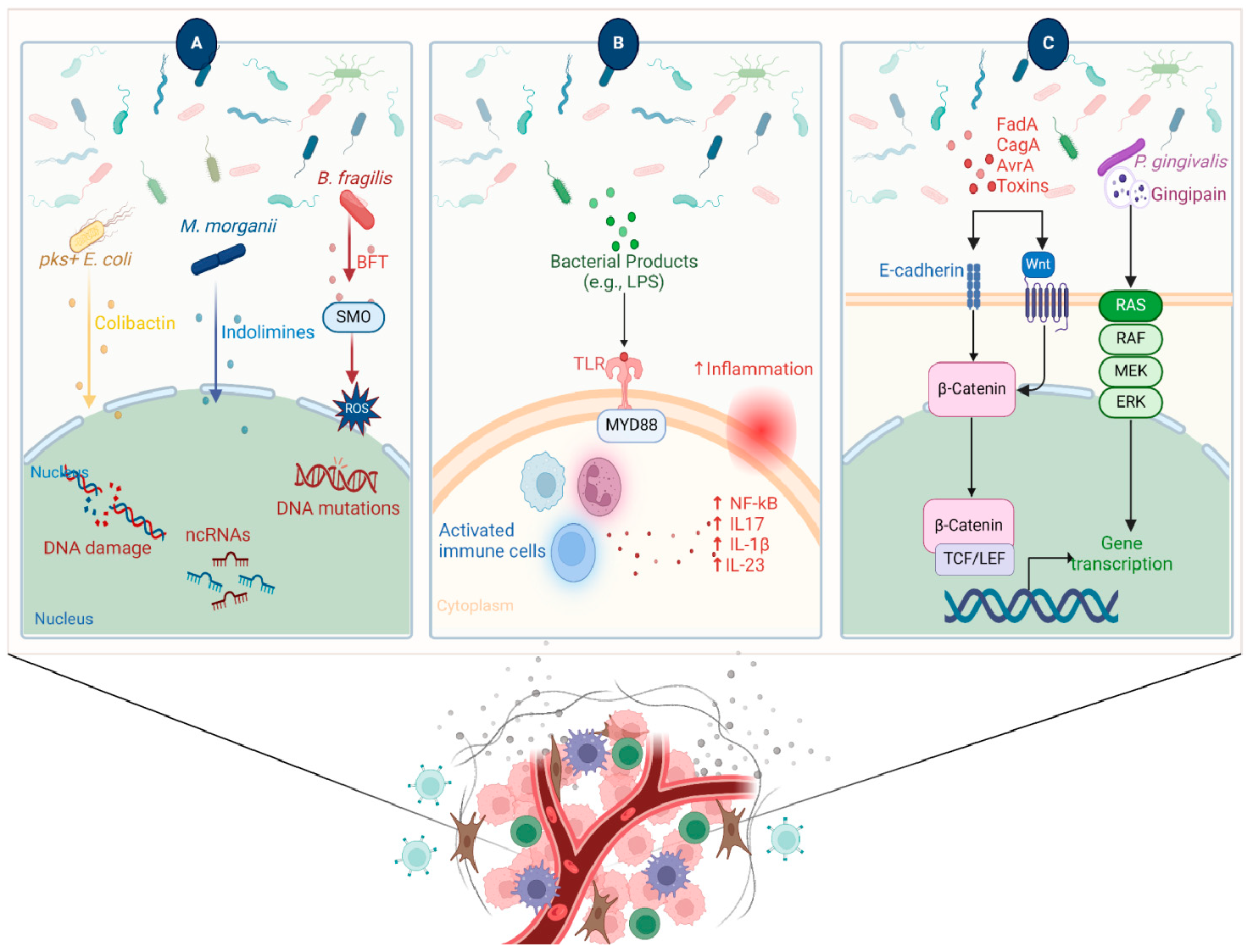
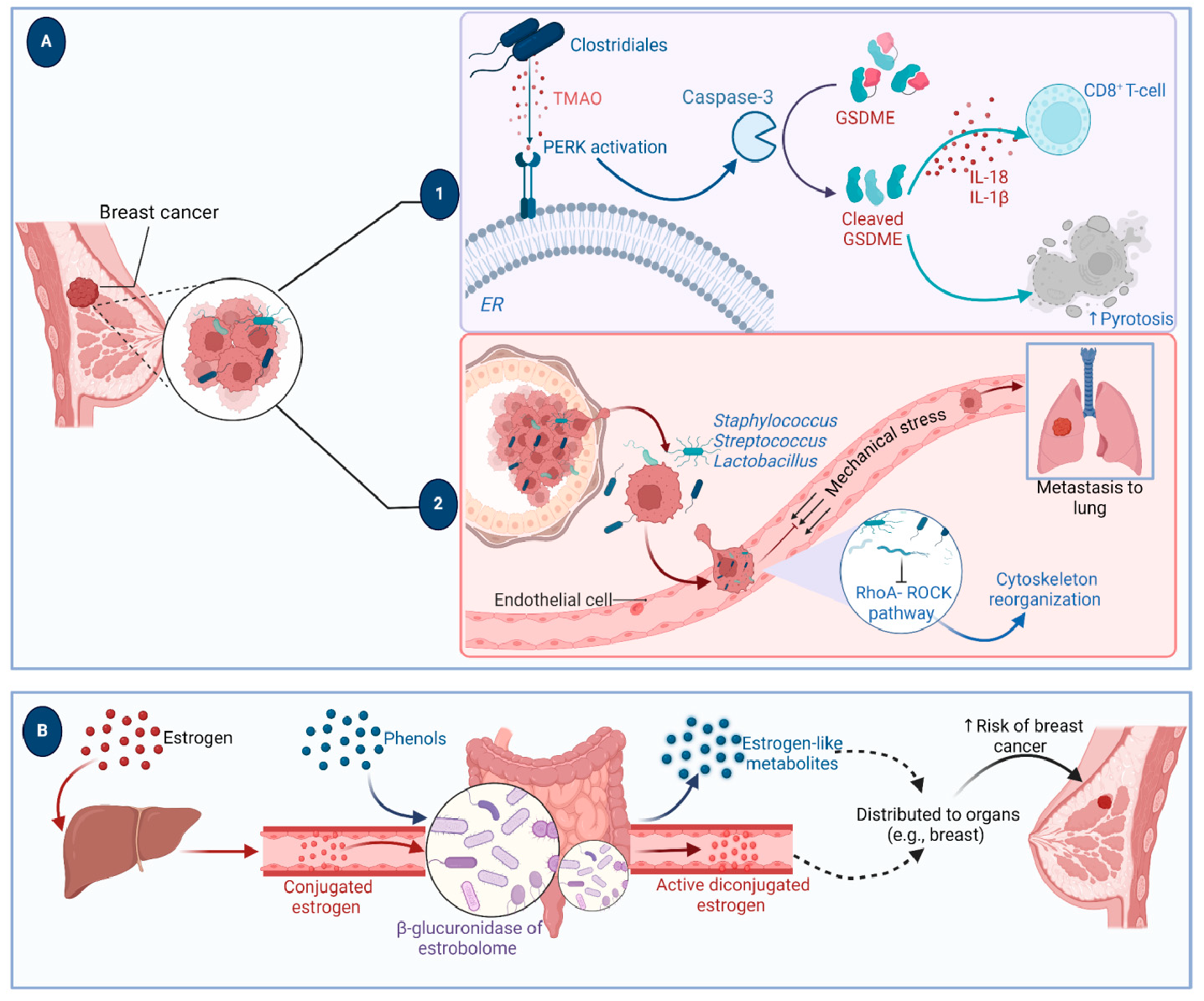
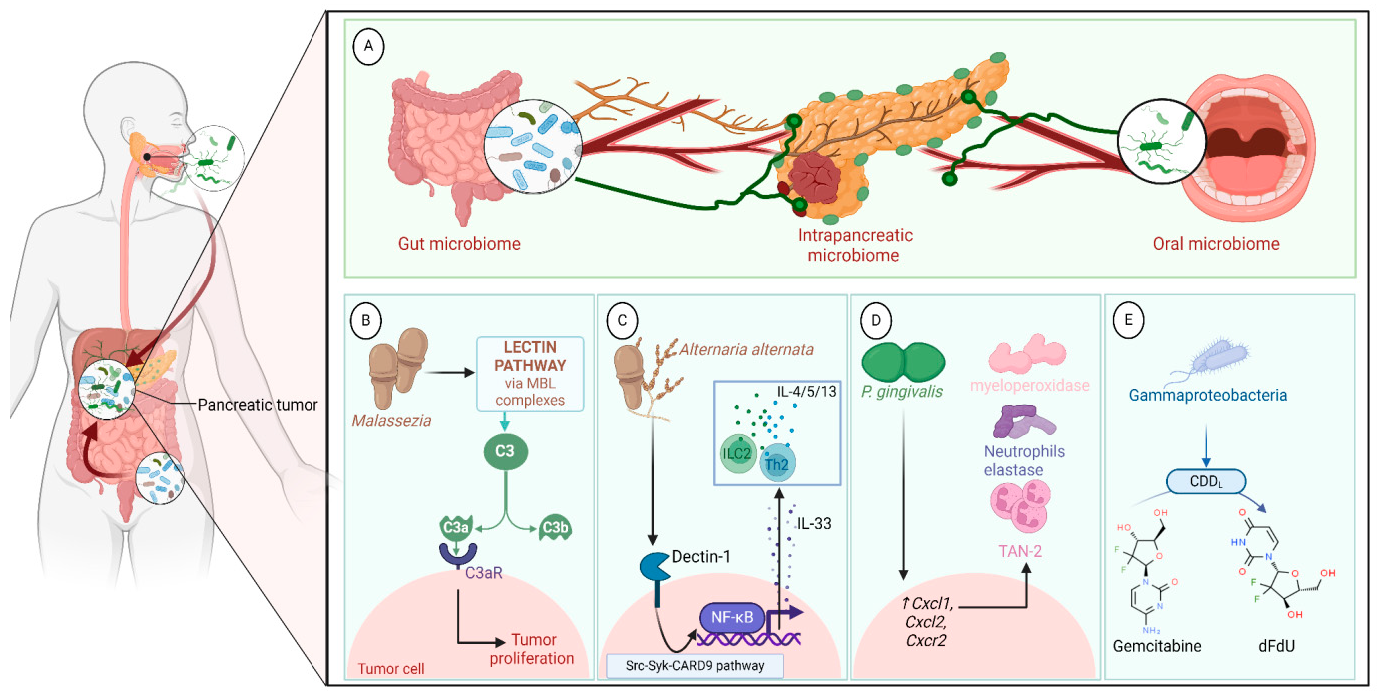
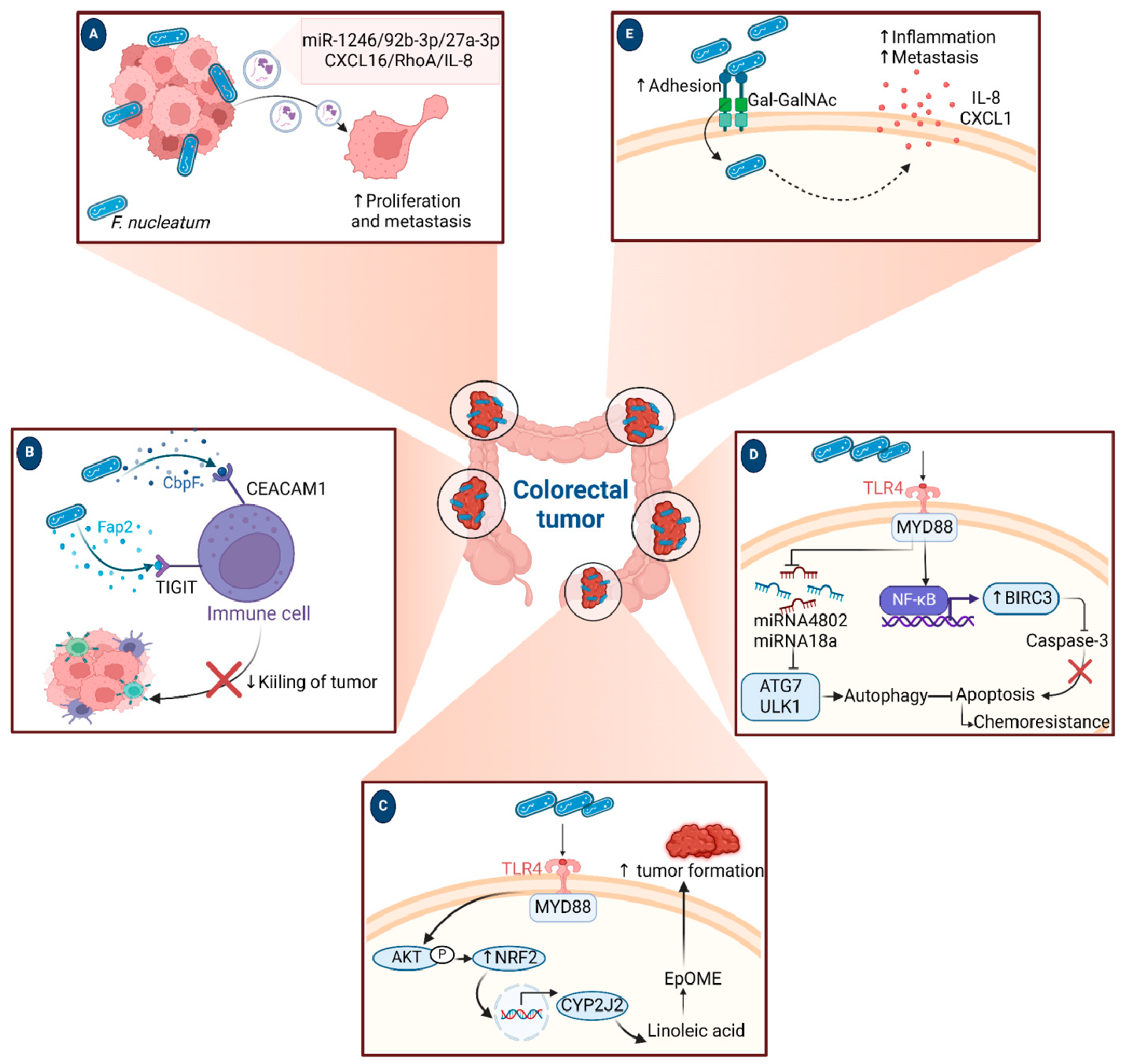
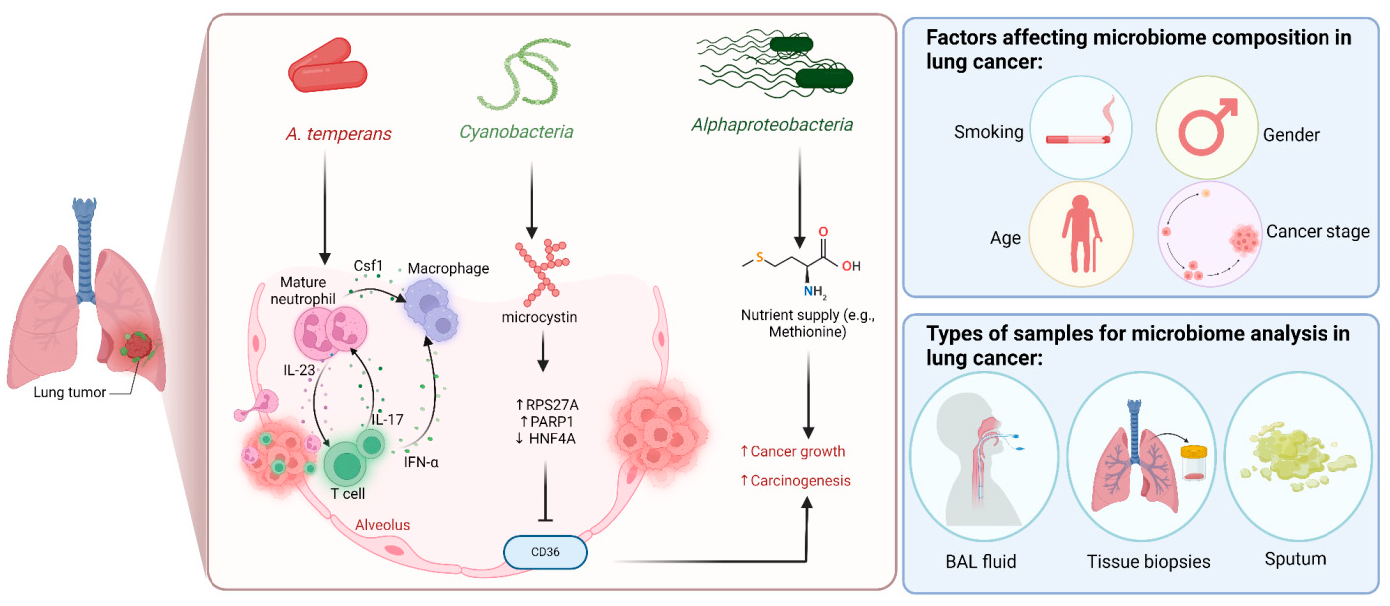
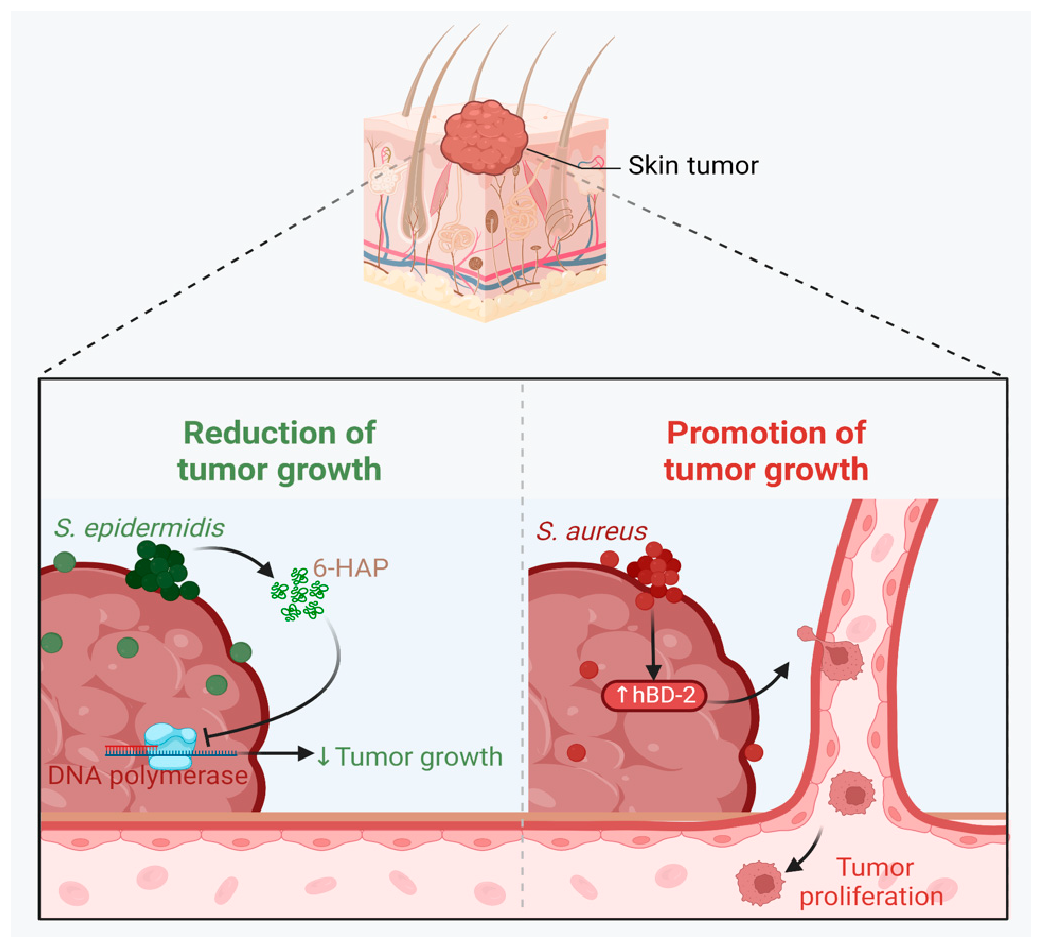
A growing body of data suggests a distinctive effect of local tumor microbiota that is independent of gut microbes. For example, in gastric cancer, the abundance of Methylobacterium inside the tumor, independent of the concentration in the feces, was found to be negatively correlated with tumor-infiltrating CD8+ T cells, downregulation of transforming growth factor-β (TGF-β) [69]. In line with this finding, another study reported the presence of a unique microbial signature composed of Acinetobacter, Pseudomonas, Rhodococcus, Sphingomonas, Brevundimonas, and Ralstonia residing inside the thyroid tumor [70,71]. Another interesting study revealed that the progression of lung cancer is associated with local intratumor residents, not gut-translocated microbes. This intratumor signature is characterized by the abundance of Herbaspirillum and Sphingomonadaceae, inducing proinflammatory mediators such as IL-1β, IL-17, and IL-23. In this study, intratracheal transplant of microbes from lung tumor into mice at the initial stage of tumor development accelerated tumor progression [41]. The full characterization of the microbiome structure in the TME remains challenging due to the low biomass of these communities [72,73]. A set of guidelines for the minimum standards for conducting microbiome studies with low microbial biomass has been suggested [74]. Currently, metagenomics and proteomics analyses are widely used to facilitate the detection and identification of bacteria, depending on their DNA or their metabolites from samples directly [75]. An in-depth investigation of the intratumor microbiomes of 1526 tumor tissues across seven cancer types revealed that all tumors contain detectable levels of bacterial metabolites and genetic material, while live bacterial cells were detected residing mostly intracellularly in both tumor and immune cells [76]. The study showed that each tumor type harbors unique and distinct microbial communities, with F. nucleatum being one of the most abundant species in breast and pancreatic tumors. Colon tumors showed a high abundance of Firmicutes and Bacteroidetes, while non-intestinal tumors were enriched in Corynebacteriaceae and Micrococcaceae. Another study found an association between survival rate and signature intratumor microbiota in pancreatic ductal adenocarcinoma (PDAC) enriched in Bacillus clausii, Saccharopolyspora rectivirgula, and Streptomyces [77]. Fecal transplant from survivors to mice with pancreatic cancer increased tumor infiltration of activated CD8+ T cells and augmented serum levels of IFN-γ and IL-2, which enhanced anti-tumor immunity, while fecal transplant from short-term survivors resulted in increased tumor infiltration of Treg cells and subsequently led to an immune suppression state [77]. There are several reports identifying the oncobiome structure and unique signature in each tumor type (Table 1). The structure of the microbiome of various cancer types and its impact on cancer behavior. ↑ denotes more abundance in the tumor cells compared to healthy tissue; ↓ denotes less abundance in the tumor cells compared to healthy one; + denotes being detected in the tumor samples; HIF: Hypoxia-inducible factors/CD4 cells: Clusters of differentiation 4 cells/CXCL 1,2: C-X-C motif chemokine ligand 1,2./ccl2: chemokine (C-C motif) ligand 2. /TGF-β: Transforming growth factor-β./TRM: resident memory cells. 5.1. Breast Cancer The first study highlighting the potential pathological significance of the oncobiome in breast cancer (BC) dates back to 1971 [91]. BC exhibits a high abundance of the intratumoral microbiome [76] with significant enrichment in Proteobacteria, Firmicutes, and Actinobacteria. On average, a total of 16.4 distinct bacterial species could be detected within each sample. In contrast, it was observed that the average number of bacterial species present in all other types of tumors was found to be less than nine. BC samples were enriched in F. nucleatum in addition to other genera such as Corynebacterium US_1715, Lactobacillus iners, and Streptococcus infantis. Moreover, they investigated that different breast cancer subtypes show a distinct microbiome that is very distinct from the microbiome in adjacent normal tissue and the microbiome between cancer and normal cells [76,80]. Other studies showed that Enterobacteriaceae and Staphylococcus are more abundant in BC patients compared to healthy subjects. Bacterial isolates from these BC subjects, including E. coli and S. epidermidis, were shown to elevate the levels of phosphorylated H2AX (gamma-H2AX) in treated HeLa cells, indicating DNA damage [92]. In a study that compared the microbiome in breast skin and BC tissue under aseptic conditions, cancer tissue showed greater species richness and distinct composition. In addition, they observed demonstrable differences in the microbiome between benign and malignant tissues. Fusobacterium, Atopobium, Hydrogenophaga, Gluconacetobacter, and Lactobacillus were significantly higher in women with malignant cancer. Interestingly, some metabolic pathways were predicted to be severely suppressed in malignant cancer patients, such as glycosyltransferases, methionine and cysteine metabolism, fatty acid biosynthesis, and C5-branched dibasic acid metabolism [81]. Another study showed that the advancement of malignancy is associated with a reduction in the relative abundance of Bacteroidaceae and an increase in the Agrococcus genus, suggesting a correlation between the abundance of certain microbiota within the breast and the invasiveness of the cancer [93]. A recent study utilized the PathoChip array to reveal distinct microbiome signatures for breast cancer subtypes. The study revealed that Estrogen receptor positive (ER+) BC is the most diverse, while Triple Negative (TN) BC showed the lowest oncobiome diversity [94]. Another study revealed that TN tumors exhibited an increase in seven genera, six of which were depleted in ER+ tumors [80]. Main mechanisms summarizing the impact of breast and gut microbiomes on BC are illustrated in Figure 3. 5.2. Pancreatic Cancer Multiple studies reported similarities in the microbiome between the duodenum and pancreatic tissues [95], suggesting a possible translocation of the microbiome from the gut into the pancreas (Figure 4). A study revealed that bacterial DNA was detectable in 76% of PDAC samples, compared to 15% of control samples. Deep sequencing analysis identified Enterobacteriaceae and Pseudomonadaceae families as the most abundant families in PDAC. Interestingly, further research showed that members of Enterobacteriaceae express cytidine deaminase, which can deactivate the anticancer drug, gemcitabine [96]. On the other hand, other bacterial taxa are associated with long-term survival, such as Saccharopolyspora, Pseudoxanthomonas, B. clausus, and Streptomyces [77]. Several studies have identified some oral microbiomes, such as F. nucleatum, P. gingivalis, Tannerella denticola, and Tannerella forsythia, that cause infections such as periodontal diseases, as risk factors for developing pancreatic cancer [97,98,99,100]. This suggests a translocation of oral bacteria or diffusion of their metabolites to the pancreas. Mitsuhash et al. detected Fusobacterium species, originally resident in the mouth, in 8.8% of pancreatic cancer specimens [101]. Other studies supported the claim of colonization of F. nucleatum in pancreatic tumor tissue. Furthermore, DNA from F. nucleatum was detected in 15.5% of pancreatic tumors. Mechanistically, F. nucleatum stimulates the secretion of some CXC cytokine groups, such as CXCL1 and IL-8, further confirmed by increased expression of mRNA coded for CXCL1 and IL-8. Both types of cytokines bind to CXCR2 to promote cell migration by inducing autocrine signaling, leading to a poor prognosis [44]. An interesting study showed that while pancreatic cancer samples are enriched in A. ebreus and Acinetobacter baumannii compared to healthy subjects, this enrichment is consistently higher in males as compared to females. This indicates that microbiota can adopt different pathways in cancer progression according to gender or smoking status [102]. 5.3. Colorectal Cancer Colorectal cancer (CRC) is the second leading cause of cancer-related death after lung cancer, and metastasis is the leading cause of mortality among CRC patients [103]. Several reports support that the Fusobacterium genus is strongly associated with CRC [104,105,106], Figure 5. Interestingly, the abundance of F. nucleatum increases with advances in cancer stage [107]. Mechanistically, F. nucleatum induces inflammation and triggers the expression of oncogenic responses through its unique membrane protein FadA. FadA binds to E-cadherin, leading to E-cadherin phosphorylation and internalization of E-cadherin, subsequently activating β-catenin signaling, which triggers the overexpression of oncogenes. FadA genes were overexpressed in colon cancer tissues by 10–100 times compared to tissues from normal individuals [66]. Other studies reported the association between intratumoral F. nucleatum and specific tumor behavior, such as high-level microsatellite instability (MSI) [108], metastasis [109], treatment resistance [110] and poor survival. A study showed that F. nucleatum promotes the metastasis of CRC by activation of the ALPK1/NF-κB/ICAM1 pathway. Mechanistically, F. nucleatum stimulates Alpha kinase 1 (ALPK1) receptor, which in turn activates the NF-κB, leading to the upregulation of intracellular adhesion molecule 1 (ICAM-1). ICM1 is a cell membrane glycoprotein engaged in cell-cell communication and assists in metastasis by promoting the adhesion of CRC cells to endothelial cells [109]. Kong et al. postulated the ability of F. nucleatum to initiate TLR4 signaling, thereby inducing the upregulation of CYP2J2 expression within cells. Subsequently, this increased expression facilitates the catalysis of linoleic acid, resulting in the production of a larger quantity of the 12,13-epoxyoctadecenoic acid (12,13-EpOME) metabolite. This metabolite contributes to the initiation of epithelial-mesenchymal transformation (EMT), a process closely associated with the development and progression of colorectal cancer [111]. Studying the role of F. nucleatum in CRC cell lines and mice models reveals that 50 miRNAs increased significantly, and 52 miRNAs were significantly negatively regulated. miR21 was the most up-regulated and contributes to carcinogenesis through stimulation of the TLR4-Myd88-NFkB pathway [112]. Another study reported similar findings on the role of F. nucleatum in the TLR4-Myd88-NFkB pathway. They showed that F. nucleatum can cause selective loss of miR-18a and miR-4802, which activate cancer autophagy and consequently promote chemoresistance in patients with colorectal cancer [110]. Another bacterium implicated in CRC is E. coli. A study showed that the detection of E. coli within colorectal biopsies is 20% in the mucosa of healthy individuals compared to 55% in CRC patients [113]. Some strains of E. coli might contribute to CRC by producing the genotoxin colibactin [55]. Campylobacter is another genotoxin-producing bacterium that is enriched among CRC patients [114]. Similar to E. coli, Campylobacter is associated with host DNA double-strand breaks [114]. CRC patients with a high abundance of Campylobacter show a mutational signature and genetic alterations such as HRAS, TSC2, AR, FGFR3, and AKT1 [115]. Metatranscriptomic analysis revealed other dominant gram-negative anaerobic bacteria among 65 cohorts. Leptotrichia and Campylobacter spp. are enriched in CRC. This signature composition (F. nucleatum, Leptotrichia, and Campylobacter) has been linked to the overexpression of certain genes in the CRC host, such as IL-8 and cathepsin Z. [116]. Other studies reveal a microbial signature characterized by a higher abundance of the Coriobacteridae subclass (Slackia and Collinsella), together with a lower abundance of Enterobacteriaceae (Kluyvera, Citrobacter, Serratia, Cronobacter, Shigella, and Salmonella spp.) in CRC [117]. 5.4. Gastric Cancer Gastric cancer (GC) is the fifth most prevalent malignant cancer and is ranked as the fourth leading cause of cancer-related mortality [118]. Studies revealed that the GC microbiota has lower microbial diversity with enrichment in Oceanobacter, Syntrophomonas and Methylobacterium genera [69]. Furthermore, Methylobacterium levels are inversely correlated with CD8+ TRM and TGFβ in TME. [69]. However, the mechanism by which Methylobacterium suppresses TGFβ is not understood. Besides, a higher abundance of Propionibacterium acnes, primarily found within the skin, was found in stage III of GC tissues than in stages I and II. P. acnes stimulates the M2 polarization of macrophages through TLR4/PI3K/Akt signaling, leading to overexpression of IL-10 [119]. H. pylori (HP) infection is among the major risk factors for GC [120,121] Approximately 70% of GC patients were diagnosed as HP+, while the eradication of HP could be a preventive measure for GC [122,123]. However, HP shows a decreased relative abundance inside gastric tumor tissues compared to normal tissues [122,124] suggesting that HP might have a role in driving chronic inflammation, enabling GC initiation, but not as an intratumor resident. HP infection activates NF-ĸB in bile duct carcinoma cells, thereby increasing expression of VEGF, a major angiogenic factor. Additionally, VEGF may elevate nuclear expression of E2F, which increases proliferation in bile duct carcinoma [125]. 5.5. Lung Cancer Lung cancer (LC) is the leading cause of cancer deaths despite the huge advances in detection methods and treatment availability. Pulmonary infection and dysbiosis of the lungs are linked to many respiratory disorders, including LC, mainly via triggering a state of chronic inflammation [126]. This occurs by stimulating Myd88-dependent IL-1β and IL-23 production from myeloid cells, consequently leading to the activation of lung-resident γδ T cells producing IL-17 and other effector molecules that promote inflammation and stimulate tumor cell proliferation [41]. However, due to ethical considerations, obtaining lung biopsy samples from healthy human subjects is not applicable. Therefore, the majority of studies used bronchoalveolar lavage (BAL) [127], sputum [128], or bronchoscopic brushing [129] to study lung microbiota (Figure 6). In one investigation involving BAL fluid in LC patients, a notable rise in abundance was observed in two phyla, namely Saccharibacteria (TM7) and Firmicutes, as well as four genera, Selenomonas, Atopobium, Megasphaera, and Veillonella. [127]. Another study linked the higher level of chromosomal aberrations in LC patients with a higher sputum abundance of Lachnoanaerobaculum, Bacteroides, Mycoplasma, Porphyromonas, and Fusobacterium in their sputum [128]. To investigate if LC microbiome composition differs according to the type of sample, a study conducted by Bingula et al. characterized the lung microbiota from three different lung tissues (tumor tissue, peritumoral tissue, and non-malignant tissue) and compared it with BAL (obtained directly on an excised lobe) and saliva samples. The microbiome in the oral and lung shows differences in diversity and taxonomy. Lung tissue samples were predominantly with Proteobacteria. While saliva and BAL samples show a high abundance of Firmicutes. However, the dominant class among saliva was Bacilli, whereas Clostridia was the dominant class among BAL samples [130]. In the same way, Patnaik et al. identified variations in the microbiome between tissue, BAL, and saliva samples [131]. This indicates the importance of sample sources to analyze lung microbiota, and it is essential to note that BAL fluid, sputum, or saliva may not precisely represent the lung microbiota due to the potential contamination of the upper respiratory tract or oral microbiota. A recent study investigated the association between intratumoral microbiome in non-small cell lung cancer (NSCLC) patients without lung infection and various factors such as malignancy, response to first-line treatment, and survival. Serratia marcescens- and Enterobacter cloacae-rich tumors were more likely to metastasize to the brain and mediastinal lymph nodes, respectively. Furthermore, Haemophilus parainfluenzae was negatively correlated with response to the first-line treatment for stage IV lung cancer; consequently, it was related to poor progression-free survival (PFS) while S. haemolyticus was linked to longer PFS [132]. Gammaproteobacteria were linked to low PD-L1 expression and poor response to checkpoint-based immunotherapy, translating into poor survival [79]. Additionally, the association of six bacterial biomarkers (Clostridioides, Shewanella, Succinimonas, Acidovorax, Dickeya, and Leuconostoc) with survival in patients with lung cancer indicated their potential to identify recurrence or metastasis [133]. By applying RNA-seq to investigate the metatranscriptome of human lung cancer, Chang and colleagues identified nine enriched bacteria in lung cancer. These nine species were correlated with a low overall survival among patients with LC. Moreover, the presence of two bacterial species, Mycobacteroides franklinii and B. megaterium, was associated with high levels of CD4+ T cells and Th2 cells, respectively. This suggests that these two bacteria can play an important role in the carcinogenesis process of LC [78]. Lung microbiome is also associated with the prognosis of lung cancer. Microbial composition differences were noted according to the cancer stage. The advanced-stage lung cancer group is enriched with the genera Staphylococcus, Burkholderia, Caballeronia, Paraburkholderia, and Peptoniphilus [134]. Recent studies have revealed significant variations in the microbiota based on histopathological types of lung cancer. For example, differential abundances were observed within the NSCLC subtypes. The abundance is significantly higher in adenocarcinoma (ADC) compared to squamous cell carcinoma (SCC). Cyanobacteria can produce a toxin called microcystin, which increases the expression of Poly [ADP-ribose] polymerase 1. Through the CD36 receptor, Poly [ADP-ribose] polymerase 1 can activate inflammatory pathways, thereby contributing to inflammation-associated lung carcinogenesis [135]. Similarly, in another study, microbiome profiles in BALF showed higher microbial diversity in SCC compared to the microbiota in ADC, in which Acinetobacter, Brevundimonas, and Propionibacterium were more enriched in ADC. In contrast, Enterobacter was more enriched in SCC [136]. Among smokers, colonization of bacteria that degrade cigarette smoke metabolites, such as nicotine, phenolic compounds, toluene, and anthranilate, is higher compared to non-smokers and lung cancer patients [76,137]. Furthermore, the abundance of Adinovorax temperans was higher in smoker LC patients compared to non-smoker LC patients. Smoking, together with TP53 mutation, was linked to impairment in epithelial function, which may facilitate the invasion of carcinogenesis bacteria such as A. temperans [18]. On the contrary, Acidovorax was more abundant among non-smokers in a Chinese study conducted recently. However, enrichment of polycyclic aromatic hydrocarbon-degrading bacteria such as Massilia and Sphingobacterium was observed. Both studies reported the link between TP53 mutations, smoking, and the presence of the oncobiome [138]. 5.6. Brain Cancer Less data is available regarding the role or abundance of the microbiome in brain tumors. Recently, a study differentiated between microbial community composition in glioma tissues versus adjacent normal brain tissues by utilizing transcriptome sequencing and metabolomics, supported by an animal model, bacterial RNA and LPS were found within glioma tissues. Six genera were found to be significantly enriched in glioma tissues compared to their adjacent normal brain tissues, including Fusobacterium, Longibaculum, Intestinimonas, Pasteurella, Limosilactobacillus, and Arthrobacter. Moreover, results from animal studies revealed that F. nucleatum promoted glioma growth by increasing the levels of N-acetylneuraminic acid and the expression levels of CCL2, CXCL1, and CXCL2. Several significantly abnormal metabolic pathways were found in glioma samples, such as several amino acid metabolisms, nitrogen metabolism, and aminoacyl-tRNA biosynthesis [21]. 5.7. Liver Cancer Liver cancer is the sixth most diagnosed cancer and is the third leading cause of death among cancer-related mortality. An estimated 1.3 million people will die from liver cancer in 2040 by an increase of 56.4% compared to 2020. Hepatocellular carcinoma (HCC) represents 80% of primary liver cancer cases [139]. The alteration of normal gut microbiota increases the permeability of the gut, which leads to liver exposure to many microbial products [140]. For example, LPS-producing genera increased in early HCC patients compared to normal subjects. LPS binds to TLR4, which directly promotes HCC [141,142]. This suggests the gut microbiome as a target to prevent HCC [143]. A higher abundance of Actinobacteria was observed in HCC tissues, whereas Deinococcus-Thermus was significantly enriched in normal tissues. Additionally, Methylobacterium and Akkermansia emerged as significant prognostic markers for both overall survival (OS) and recurrence-free survival (RFS) [83]. Song et al. developed a microbiome-related score (MRS) model. This model identifies a 27-microbe prognostic signature of microbial abundances related to OS and disease-specific survival (DSS) in patients with HCC. The MRS model can predict prognosis, particularly 1-, 3-, and 5-year OS and DSS rates of HCC patients. Among the 27 microbes, some genera such as Ornithinimicrobium, Caldimonas, Holophaga, and Rheinheimera are associated with decreased overall response (OR), while others such as Robinsoniella, Snodgrassella, Amycolatopsis, Alicyclobacillus, and Tetragenococcus are linked to increased overall survival (OS) in HCC patients [144]. 5.8. Cervical Cancer A study linked the presence of L. iners in cervical tumors to treatment resistance and decreased patient survival. The Lactobacilli genus in general utilizes carbohydrates and uses lactate dehydrogenase (LDH) to produce lactate as the final product of fermentation [145]. However, L. iners does not express the D-LDH gene, and only L-lactate enantiomers are produced. Interestingly, L-lactate production increases after exposure of cells to metabolic stress such as ionization radiation. Lactate can provide energy to the tumor cells and contribute to communication between tumor cells and surrounding cells. Furthermore, lactate can activate certain signaling pathways that contribute to treatment resistance, such as hypoxia-inducible factor 1 (HIF-1) transcription targets and ROS-induced cellular signaling [146]. 5.9. Skin Cancer The main phyla of normal skin tissue are Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes [147], with the most represented genera being Corynebacteria, Propionibacteria, and Staphylococci [148]. Kullander et al. reported that the higher prevalence of S. aureus is associated with skin SCC, but not basal cell carcinoma, compared to healthy skin by analyzing tumor biopsies and swab samples. However, whether S. aureus influences carcinogenesis or if SCC has an increased susceptibility to S. aureus colonization still needs more investigation [86]. Furthermore, S. aureus overabundance was also significantly linked to increased human beta defensin-2 (hBD-2) expression in SCC samples (Figure 7). The challenge of SCC cells directly with hBD-2 promoted keratinocyte tumor cell proliferation [149]. Some studies suggest that skin damage promotes the opportunity for S. aureus to infect the skin and secrete its virulence factor, regulated by the staphylococcal accessory regulator (SarA) protein. These virulence proteins induce chronic inflammation, consequently leading to skin cancer development [150]. On the other hand, in cell culture experiments, Nakatsuji et al. identify a skin commensal microbe, S. epidermidis, that can produce 6-N-hydroxyaminopurine (6-HAP). This molecule works as a DNA polymerase inhibitor that blocks the proliferation of tumor cells. Moreover, treating mice models with 6–HAP–producing S. epidermidis reduced the incidence of UV-triggered tumors compared to control mice. Consequently, these results suggest the role of skin commensals in protection against skin cancer [151]. Furthermore, a mouse study showed that the growth of melanoma cells was inhibited upon intratumoral administration of the commensal P. acnes. The proposed mechanism was through the induction of Th1-type cytokines such as IL-12, TNF-α, and IFN-γ. Moreover, they found that the induction of IFN-γ promotes cytotoxic effects by activating CD8+ T cells, NK cells, and B cells and elevates chemokines, including CXCL9 (MIG) and CXCL10 (IP-10) that suppress vascular proliferation [152]. 5.10. Genitourinary Cancers 5.10.1. Prostate Cancer Analysis of prostate tumor specimens from 242 patients revealed that microbes were more abundant in tumor samples than in normal samples [153]. Findings from another study suggest that 70% of bacterial genera detected in prostate tumor samples were gram-negative bacteria, in which Proteobacteria were the most abundant, followed by Firmicutes, Actinobacteria, and Bacteroides. Additionally, DNA from H. pylori, specifically the sequences of the cagA gene, was detected in specific host chromosomes in prostate tumor cells. The cagA gene encodes for the immune-dominant cagA virulence factor [64]. Moreover, P. acnes infection was positively associated with chronic inflammation of the prostate. Consequent to P. acnes infection, the body activates transcription factors NF-ĸB and STAT3 that induce plasminogen-matrix metalloproteinase and COX2-prostaglandin pathways activation, leading to chronic inflammation. Prolonged exposure to P. acnes not only affects host cell proliferation but also induces cellular transformation [154]. 5.10.2. Ovarian Cancer Tissue samples from ovarian cancer showed different bacterial, fungal, viral, and parasitic characteristics in comparison with normal samples [155]. Evidence suggests a significant decrease in both the total number and diversity of bacterial communities in ovarian cancer tissues compared to those in normal distal fallopian tube tissues. Moreover, inflammation-associated signaling pathways, such as cytokine–cytokine receptor interaction, NF-κB signaling, and chemokine signaling, were significantly activated in ovarian cancer tissues [90]. 5.10.3. Bladder Cancer Comparing microbiome composition in urine and tumor tissue in bladder cancer patients revealed similarity in phyla levels, where both sample types showed Firmicutes, Proteobacteria, Actinobacteria, Cyanobacteria, and Bacteroidetes as the most abundant phyla. However, in terms of genera, urine samples were enriched in Lactobacillus, Staphylococcus, Streptococcus, and Corynebacterium. Whereas, Akkermansia, Bacteroides, Klebsiella, Enterobacter, and Clostridium sensu stricto are abundant in tissue samples [156]. Another study showed that genes of EMT, including TWIST1, E-cadherin, SNAI2, SNAI3, and vimentin, are associated with the presence of butyrate-producing bacteria [89]. 5.10.4. Kidney Cancer The kidney microbiome is originally translated from the gut, circulatory system, or ascended from the lower urinary tract [87]. It has been observed in a study that species diversity was decreased in renal cell carcinoma (RCC). In addition, a noted reduction in Streptophyta was observed in tumor tissue compared to healthy. Of note, 9 KEGG pathways were significantly different between the two groups. For example, membrane transport, transcription, and cell growth and death pathways were abundant in tumor tissues, whereas the other 6 pathways, such as energy, cofactors, and vitamins metabolism, and cell motility, were abundant in normal tissues [87].Table 1:
Cancer Type
Study Design
Sample Size
Intratumor Microbes
Outcomes
Mechanism of Action
References
lung cancer
Meta transcriptomics pilot study
49
↑
Brevundimonas diminuta,
↑
Acinetobacter radioresistens
↑
Enterobacter cloacae
↑
Mycobacterium chelonae
↑Mycobacterium franklinii
↑
Staphylococcus sp.
↑
Bacillus megaterium
↑
Pseudomonas aeruginosa
↑
Rhodococcus erythropolis
The development of cancer progression and metastasis leads to a poor prognosis.
Unknown mechanism of inducing carcinogenesis.
[78]
Prospective observational study
38
↑ Gammaproteobacteria
Lower response to anti-PD-L1
Reduced survival rate by worsening the recurrence-free survival (RFS) and overall survival (OS) rate.By lowering programmed death-ligand 1 (PD-L1) expression on cancer cells.
[79]
Breast cancer
(BC)
Cross-sectional study
221
↓Streptococcus
↓Propionibacterium
↓Anaerococcus,
↓Caulobacter
↓Streptococcus
↑Porphyromonas
↑Lacibacter
↑Ezakiella,
↑FusobacteriumEnhancing tumor suppression
- Streptococcus and Propionibacterium activate an anti-tumor response by activating T-cells.
[80]
Cross-sectional study
33
↑
Gluconacetobacte
↑Fusobacterium
↑Atopobium,
↑Lactobacillus
↑Hydrogenaphagar
Stimulating tumor progression and metastasis.
Creating a proinflammatory environment and secreting virulence factors that induce carcinogenesis.
[81]
Pancreatic cancer
In vivo/in vitro study
125
↑Fusobacterium nucleatum
Induction of pancreatic tumor growth and metastasis, leading to poor prognosis.
- Promoting the secretion of motif chemokine ligand 1(CXCL1), which will activate the autocrine signaling pathway.
- Modifying the tumor microenvironment (TME) by suppressing the activity of the infiltrating tumor CD8+ cells.[44]
Retrospective cohort study
68
+Pseudoxanthomonas +Streptomyces
+Saccharopolyspora
+Bacillus clausii
Enhancing the therapy outcomes, as they were found to be more abundant in long-term survival patients.
Activating and recruiting CD8+ immune cells to the tumor cells.
[77]
Liver cancer
Retrospective analysis
28
↓Pseudomonadaceae
↑
Rhizobiaceae
↑
Agrobacterium
- Peudomonadaceae: exerts anti-tumor effect and acts as an effective therapeutic agent.
- High abundance of Rhizobiaceae and Agrobacterium in the cancer cells may be associated with tumor progression.Unknown mechanisms
[82]
Retrospective analysis
91
↑ Proteobacteria
↑Actinobacteria,
↓Deinococcus thermus.
↑Akkermansia
↑Methylobacterium
Proteobacteria & Actinobacteria: increase pathogenesis and tumor progression.
Akkermansia and Methylobacterium: acting as effective predictors for better recurrence-free survival (RFS) and overall survival (OS).Proteobacteria: It is involved in the pathogenicity of endotoxemia and inflammation.
Actinobacteria: highly present in patients with poor prognosis.[83]
Cervical cancer
Retrospective analysis
72
↑ Klebsiella
+Micromonospora
+Microbispora
+Methylobacter
Induction of metastasis and tumor progression.
Increase the production of expression of HIF-mRNA in the epithelial cells, causing epithelial-mesenchymal transition.
[84]
Colorectal cancer (CRC)
Multi-omics analysis
372
+ Clostridium
+ Flavonifractor
+ Parvimonas micra
+ Fusobacterium nucleatum
+ Alistipes
+ Oscillibacter
+ Akkermansia
- Clostridium, Fusobacterium nucleatum: confer a more malignant phenotype to CRC cells and promote colorectal tumorigenesis and metastasis.
- Akkermansia:
increase therapy response.
- Parvimonas micra: contribute to tumorigenesis.
- Odoribacter splanchnicus: protection against tumorigenesis.
Flavonifractor: negative correlation with survival time.- Akkermansia: modulates the tumor microenvironment (TME) and activates immune cells like t- T-cells and natural killer (NK) cells.
- Odoribacter splanchnicus:- induce intestinal th17 cells development against CRC.
- Clostridium may affect tumor-infiltrating immune cells (TIICs), particularly mucosa-associated invariant T (MAIT) cells.
- Fusobacterium nucleatum: The abundance of tumor-infiltrating M2-like macrophages will be increased.
- Parvimonas micra: It promotes differentiation of CD4+ T cells to Th17, increases the oncogenic signaling pathway.[85]
Squamous cell carcinoma (SCC)
Case-control study
353
↑ Staphylococcus aureus (S. aureus)
Promoting tumor development and progression.
Induce chronic inflammation in the skin, leading to the production of tumor necrosis factor (TNF), which will activate nuclear factor-κB (NF-κB), a transcription factor.
[86]
Brain tumor (glioma)
multi-omics study
50
↑Fusobacterium nucleatum
↑Longibaculum
↑Intestinimonas
↑ Pasteurella
↑Limosilactobacillus
↑ Arthrobacter. Contribute to tumor progression and metastasis.
F. nucleatum increases N-acetylneuraminic acid and CCL2, CXCL1, CXCL2, and chemokine expression levels.
[21]
Kidney cancer (KC)
Case-control study
24
↑ Deinococcus.
↑Rhodoplanes
↓Cyanobacteria (class Chloroplast and the order Streptophyta)Cyanobacteria restrict metastasis and tumor growth.
Deinococcus and Rhodoplanes cause cancer development.Cyanobacteria produce bioactive substances that can induce cancer cells’ apoptosis.
[87]
Gastric tumor
Mouse model
And single-cell sequencing.53
↑ Methylobacterium
Causing tumor progression and poor prognosis
- Reduction in CD8+ and Tissue-resident memory cells (TRM).
- Reduction in the level of TGF-beta in tumor microenvironment (TME), which will inhibit the production of CD103 TRM cells, leading to the tumor’s escape from the immune system.[69]
prostate tumor
Cross-sectional study
16
↑ Staphylococcus spp.
↑ Propionibacterium spp.-Increase in tumor invasiveness and progression.
-Propionibacterium spp. Able to make biofilms and adhere to the components of the extracellular matrix.
[88]
Bladder cancer
Observational study
400
+E. coli,
+butyrate-producing bacterium SM4/1
+species of Oscillatoria
Epithelial–mesenchymal transition (EMT) genes are involved in the progression and metastasis of tumors.
- important correlations between the abundance of those bacteria and 30 epithelial–mesenchymal transition (EMT) genes in bladder cancer.
[89]
Ovarian cancer
Cross-sectional study
50
↑
Ratio of Proteobacteria/Firmicutes
↑
Acinetobacter lwoffii
↓
Lactococcus piscium
High Ratio of Proteobacteria/Firmicutes
and Acinetobacter lwoffii associated with tumor progression and metastasis.
Lactococcus piscium can act as a marker for tumorigenesis absence.- Activation of the inflammation-related pathways was observed in tumor tissue samples.
- Acinetobacter lwoffii causes persistent infection and escapes the host immune system.
Lactococcus piscium acts as a microbial biomarker to distinguish between benign and malignant tissue.[90]
Several studies revealed the significant role of the intratumor microbiome in influencing the response to cancer therapy and, in particular, immunotherapeutics (Table 2). For example, the efficacy of various chemotherapeutic drugs, such as gemcitabine, fludarabine, and cladribine, could be attenuated or enhanced by bacteria commonly present in tumor tissues. This influence is, in part, mediated by bacterial modification of the chemical structure of drugs. For instance, intratumor Gammaproteobacteria, expressing the bacterial enzyme cytidine deaminase, have been linked to gemcitabine resistance in cancers, including colon and pancreatic cancer. Conversely, microbiota-derived tryptophan metabolite indole-3-acetic acid has shown promise in enhancing chemotherapeutic effects in pancreatic cancer by modulating ROS accumulation and downregulating autophagy (https://doi.org/10.1002/mco2.376). In colorectal cancer, F. nucleatum has been implicated in activating pathways, like TLR4, to enhance autophagy in cancer cells, leading to chemoresistance [157]. In prostate cancer, the intratumor LPS-activated NF–κB–IL6–STAT3 axis has been associated with increased proliferation and chemoresistance (https://doi.org/10.1016/j.cell.2017.07.008). It comes as no surprise that the significant impact of intratumor microbiota on the efficacy of immunotherapeutics, such as checkpoint inhibitors, is observed, given the crucial interaction of these microbes with the immune system. For example, a study showed that some defined taxa can improve anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), increase the accumulation of IFN-γ-producing CD8+ T cells, and improve the efficacy of anti-PD-1 therapy [158]. These taxa include Bacteroides, Ruminococcaceae, Parabacteroides, and Alistipes. Further studies showed that fecal transplant enriched with SCFAs producers increased tumor infiltration of CD8+ T cells and improved the outcome of anti-PD-1 immunotherapy in melanoma patients [159,160]. A recent study showed that a higher abundance of Ruminococcus, Bacteroides, and Faecalibacterium is associated with increased responses to CAR-T cell therapy in B-cell malignancies [161]. Another study reported two oncomicrobiotics named E. hirae and Barnesiella intestinihominis, to enhance the recruitment of IFN-γ-producing γδ T cells and CD8+ effector tumor-infiltrating lymphocytes while reducing Treg cells and γδ T17 inside tumor cells, leading to improved outcome of cyclophosphamide therapy [14]. The impact of Gut microbiome on the efficacy of Immune checkpoint inhibitors therapy. Up arrow: means increase; Down arrow: Means decrease.Table 2:
Type of Cancer
Study Size
Type of Immuno-Therapy
Sample
Outcomes
References
Melanoma
25
Anti-PDI-1
or anti PDI-1/Anti-CTLA-4)Feces
↑E. biforme, Ruminococcus gnavus, E. coli, Streptococcus salivarius, and Phascolarctobacterium succinatutens, in respondent patients.
↑B. longum, Prevotella copri, Coprococcus sp, Eggerthella, and Eubacterium ramulus in non-respondent patients.
↑Streptococcus parasanguinis carriers → longer Overall Survival.
↑B. massiliensis → higher in Progression-free survival.
↑Peptostreptococcaceae carriers → shorter overall survival and progression-free survival rate.[162]
Advanced thoracic
carcinoma
42
PD-1 blockade
Feces
↑Enterobacteriaceae, Carnobacteriaceae, Akkermansiaceae, Enterococcaceae, and Clostridiales in the respondent group correlated with a longer Progression-free survival rate.
[163]
Advanced-stage GI Carcinoma
74
Anti-PDI-1, or Anti PD-1/Anti-CTLA-4
Feces
↑Ruminococcaceae, Lachnospiraceae, and Prevotellaceae in Respondent individuals.
↓Bacteroidaceae in Respondent individuals.
Prevotella/Bacteroides ratio decreased in the respondent individuals.
Producing short-chain fatty acid (Lactobacillus, Streptococcus, and Eubacterium) → positively correlated with anti-PD-1/PD-L1 response. [164]
Hepatocellular carcinoma
8
PD-1 blockade
Feces
↑Proteobacteria abundance in non-respondents during therapy.
[165]
Non-Small Cell Lung Cancer
11
PD-1 blockade
Feces
↑A. muciniphila, B. longum, Faecalibacterium prausnitzii in Respondents.
↑Staphylococcus aureus, Veillonella, Propionibacterium acnes, Peptostreptococcus, Sutterella, Dialister, and Ruminococcus bromii in non-respondent patients.
↑Streptococcus, Lactobacillus, Enterobacteriaceae, Prevotella, Bacteroides plebeius, Oscillospira, and Rikenellaceae are present in cancer patients compared to healthy control participants.[166]
Melanoma
112
Anti-CTLA-4, Anti-PDI-1
Feces and oral samples
↑variety of alpha and relative abundance of Ruminococcaceae bacteria in respondents.
[2]
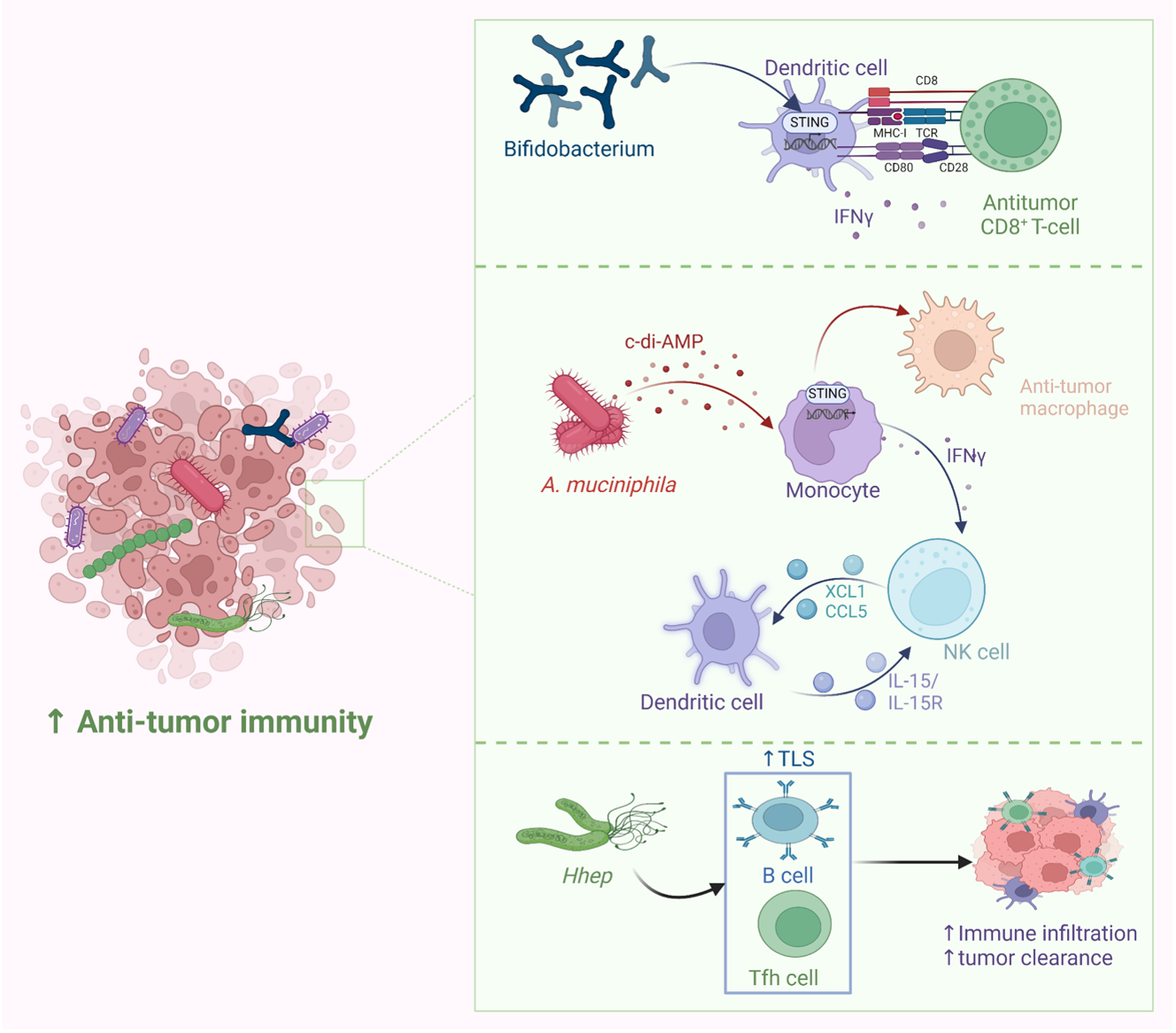
Probiotics have been widely employed to confer health benefits [167] by restoring the healthy microbiome structure and its associated beneficial functions [168,169]. Multiple studies show the beneficial effect of using specific microbes as an adjuvant with chemotherapeutics (Figure 8). For example, co-administration of Eudoraea spp. anti-PD-1 resulted in a better outcome of immunotherapy through the activation of CD8+ T cells and cytolytic T cells in the mouse model [170]. Combining Bifidobacterium with anti-PD-L1 therapy reduced tumor expansion by enhancing the activity of dendritic cells and increasing the intratumor accumulation of CD8+ T cells [171]. A study conducted on the CRC mice model fed on nano-sized L. plantarum showed a reduction in the number of tumor lesions compared to the control. These changes were attributed to the induction of cell cycle arrest and apoptosis, the suppression of inflammation, and increased IgA secretion [172]. Another study showed that the probiotic VSL#3, which is composed of Bifidobacterium and Lactobacillus species, reduced the proliferating cell nuclear antigen labeling index, TNFα, IL-1β, IL-6 production, and COX-2 expression, and increased IL-10 levels in colon tissue [173]. Emerging data suggest that the intratumor microbiome signature could be used as a diagnostic biomarker [174]. Although being technically challenging due to the difficult-to-access sampling sites, low microbial biomass, and the high chance of contamination [174]. For example, a study examining the microbiota associated with esophageal SCC revealed that patients have a reduced microbial diversity characterized by lower abundances of Bacteroidetes, Fusobacteria, and Spirochaetes. Interestingly, the authors claim that this microbial shift could effectively distinguish between patients and healthy controls. Furthermore, this dysbiosis affected the metabolic profile with a change in nitrate reductase level [175]. Another study suggests that P. somerae can be used as a biomarker for endometrial cancer. P. somerae upregulates the hypoxia-inducible factor pathway, a hallmark of endometrial cancer (10). Another study suggested that oral microbiota, such as P. gingivalis and Aggregatibacter actinomycetemcomitans, can be used to predict the possibility of developing pancreatic cancer [176]. Routy et al. reported the ability of A. muciniphila to modulate the link between immunotherapy and treatment response. Administration of A. muciniphila after the initiation of fecal microbiota transplantation using feces from non-responding mice has restored the responsiveness to PD-1 blockade, showing a promising interleukin-12-dependent mechanism [5]. Another study involving preclinical oral probiotics in mice with bladder cancer and melanoma showed that administering Bifidobacterium will enhance the tumor control significantly when combined with PD-L1 blockade [171]. Le Noci et al. reported that L. rhamnosus could enhance the immunosuppression reversal and inhibitory effect of lung tumor implantation, while also further reducing the number of metastases when alternating with antibiotics. Together, they show that the microbiota of the local environment seems to play key roles in the immune response and its implication in lung cancer [177]. Another study investigated the influence of intratumor microbiota on CD47-based cancer immunotherapy in colon cancer. Shi et al. administered Bifidobacterium to colon cancer patients, and found that it has been colonized and accumulated inside tumor sites, resulting in the augmentation of local anti-CD47 treatment via a STING-dependent route [178]. Moreover, Iida et al. showed that administering Alistipes shahii via oral gavage was sufficient for bringing back the immunotherapeutic response against colon tumors in mouse models, which had been treated previously with antibiotics [3].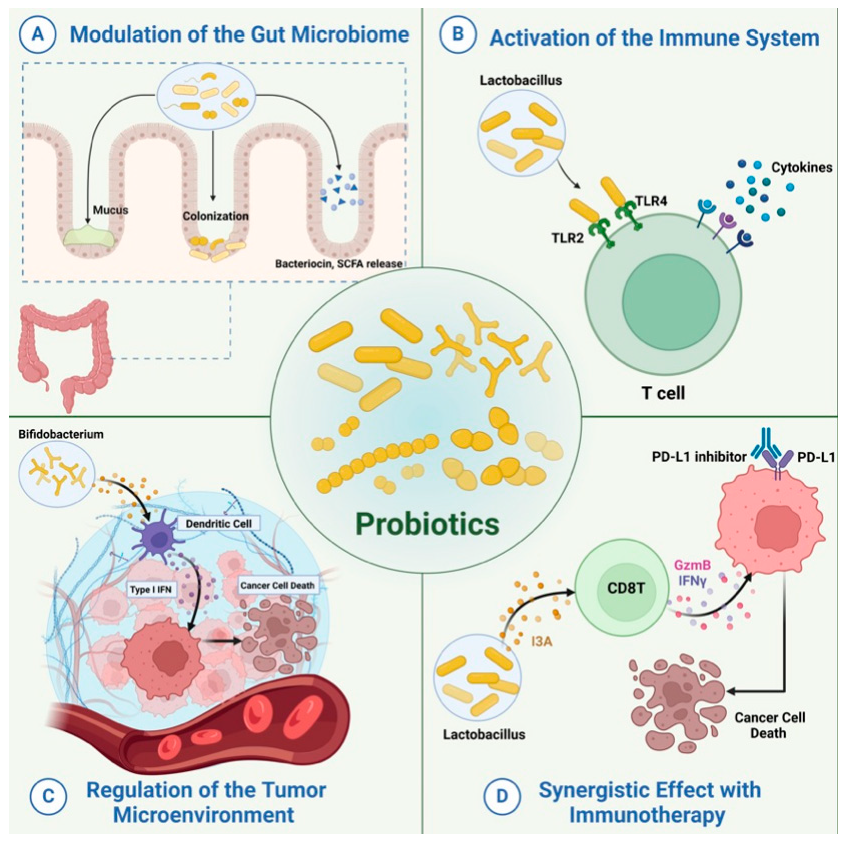
The design and development of engineered or programmed probiotics for treating a range of human conditions, from inflammatory bowel diseases to cancer, is gaining momentum [168]. This interest is fueled by the advancement in gene editing technology, including third-generation Clustered Regularly Spaced Short Palindromic Repeats (CRISPR)/CRISPR-associated Protein (CRISPR-Cas) system [179]. Engineered probiotics are modified microorganisms that can deliver a more controlled outcome compared to conventional probiotics, with unpredictable interactions within the host context [180]. The application of engineered probiotics in cancer therapy includes their use as; (1) adjuvant therapy to enhance the efficacy of immunotherapeutics, (2) heterologous host to express anticancer drugs, (3) vectors to ensure the precise delivery of anti-tumor drugs, and (4) a non-invasive technique to sense and detect tumor cells. Examples of bacteria highly utilized in engineered probiotics include E. coli, Bifidobacterium, and S. typhimurium. These microbes are anaerobes that can easily survive, effectively colonize, and carry anticancer proteins, drugs, and compounds to the intratumor anaerobic environment. E. coli Nissle 1917 (EcN) is one of the most utilized strains in the field of engineered probiotics, due to its well-known tolerability in humans and high safety margin, in addition to being easily genetically manipulated [181,182]. Various studies have been carried out in this regard, utilizing in vitro cell lines, in vivo models, and clinical trials, to prove the efficacy and safety of such living biotherapeutic products (LBP) [183]. Data indicates that metabolic modulation of the intratumor environment via engineered probiotics can act synergistically with other immunotherapy agents to achieve durable and potent eradication of cancer [184]. Examples of the use of engineered probiotics in cancer therapy or diagnosis are detailed (Figure 9). Many reports highlight the promise and efficacy of engineered probiotics in provoking anti-tumor activity and enhancing the activity of cancer therapy. For example, an engineered strain of S. typhimurium expressing IL-15/Flagellin B (FlaB) proteins causes tumor regression in animal models of metastatic colon tumors [185]. FlaB is a protein used as an adjuvant in vaccines due to its strong ability to activate the innate immune response, primarily by enhancing the recruitment of immune cells [186,187]. IL-15 has immunostimulatory action mainly by promoting maturation, development, and activation of NK, NKT, and CD8+ cells, and increasing proliferation of the specialized CD8+ T memory cells [188,189]. Engineered S. typhimurium induces both the innate and adaptive immune response, suppressing tumor growth in mice and enhancing the development of immune memory toward the tumor cells. Besides that, the combination of engineered S. typhimurium producing IL15/FlaB and PD-L1 blockade treatment revealed an improved efficacy of this synergistic combination, including in metastatic cancers [185]. Another engineered probiotic strain, EcN, was developed to constantly convert the tumor byproduct ammonia to L-arginine, a key element in provoking the immune response, mainly through increasing the proliferation of T cells. The use of this engineered probiotic increased the number of tumor-infiltrating T cells, resulting in the suppression of tumor growth in the MC38 tumor model when combined with PD-L1 antibodies. Additionally, mice injected with the EcN-engineered strain were found to form T cell memory specifically against MC38 tumors, which yields long-term protection [190]. Engineered B. longum (BL) was developed to express tumstatin, a potent angiogenesis inhibitor. Tumstatin-transformed BL exerted significant anti-tumor activity, supported by a reduction in the volume, weight, growth, and microvessel density of the tumors. Also, the intratumorally expressed tumstatin generated apoptosis and stimulated the immune response toward tumor cells. The implication of the Tum-BL system is expected to gain momentum when thinking about new approaches to treat solid tumors [191]. Interestingly, some engineered probiotics have reached clinical trials, such as SYNB1891, an engineered EcN developed by Synlogic (NCT04167137) [192]. SYNB1891 activates antigen-presenting cells, triggering innate immunity in addition to stimulation of the interferon pathway through the production of di-AMP. Multiple studies showed the efficacy of engineered probiotics in the targeted delivery of therapeutics inside the tumor cells. For example, E. coli SLIC was engineered to deliver checkpoint inhibitors such as PD-L1 and CTLA-4 antagonists in the form of nanobodies and control their release inside the tumor cells utilizing a stabilized lysing release system that was optimized based on computational and experimental studies. Data shows that a single intravenous or intratumor injection of such a system resulted in a higher therapeutic response compared to antibodies, resulting in restriction of tumor growth in mice. The authors suggested that this activity is mediated by a systemic stimulation of the immune response, as suggested by the increased number of T cells [193]. Another study employed tumor tropism to enable guiding the bacteria to tumor cells. An example is EcN, an engineered E. coli Nissle 1917, which is designed to deliver tumor suppressors such as tumor suppressor p53 and the angiogenic inhibitor TUM-5 to the tumor sites. Use of this engineered strain resulted in restricted tumor growth in mice [194]. Data shows that EcN can accumulate inside the hypoxic tumor microenvironment in nude mice. Blue light is employed to control the expression of specific TNFα in the genetically engineered EcN (EcN@EL222-TNFα). Such strain was modified to be sensitive to the applied blue light and accordingly produces TNFα in tumor tissues. Specialized nanoparticles were subsequently injected following the delivery of engineered blue-light sensitive E. coli strain, to accomplish the key role, which is the conversion of near-infrared light (NIR) that originates from a laser light applied exogenously, to a local blue light, resulting in a direct illumination endogenously toward the specific EL222 in the E. coli strain, stimulating it to produce TNFα. As long as laser light is shed from outside, the engineered probiotic will continue to produce necrosis factor from inside, and once removed, the whole process of TNFα expression will stop. The results exhibit a considerable efficacy of NIR light-responsive E. Coli strain to inhibit the tumor growth both in vitro against stage IV human breast cancer cell lines, and in vivo using mouse models. This provided a valuable approach for the precise regulation of intratumor drug delivery [195]. Another emerging application of engineered probiotics is the sensing and diagnosis of tumors. The TME is attractive to anaerobic microbes such as E. coli and Clostridium EcN named PROP-Z, which was designed to selectively detect liver metastasis in mice. PROP-Z is engineered to co-express luciferase and β-galactosidase and thus can generate luminescent and colorimetric signals [196]. Oral treatment of PROP-Z coupled with intraperitoneal injection of D-luciferin resulted in a detectable tumor-specific signal in the urine that is proportional to the size of the tumor in a murine model of liver metastasis. A further modification that included the introduction of the gene dlp7 from B. subtilis and a toxin-antitoxin system has further enhanced the efficacy and stability of the construct. Oral administration of PROP-Z and a combined luciferin/galactose molecule, named LuGal, resulted in the release of luciferin by the action of β-galactosidase. luciferin is then detected in the urine. Interestingly, this programmed strain was not able to colonize healthy tissues [197].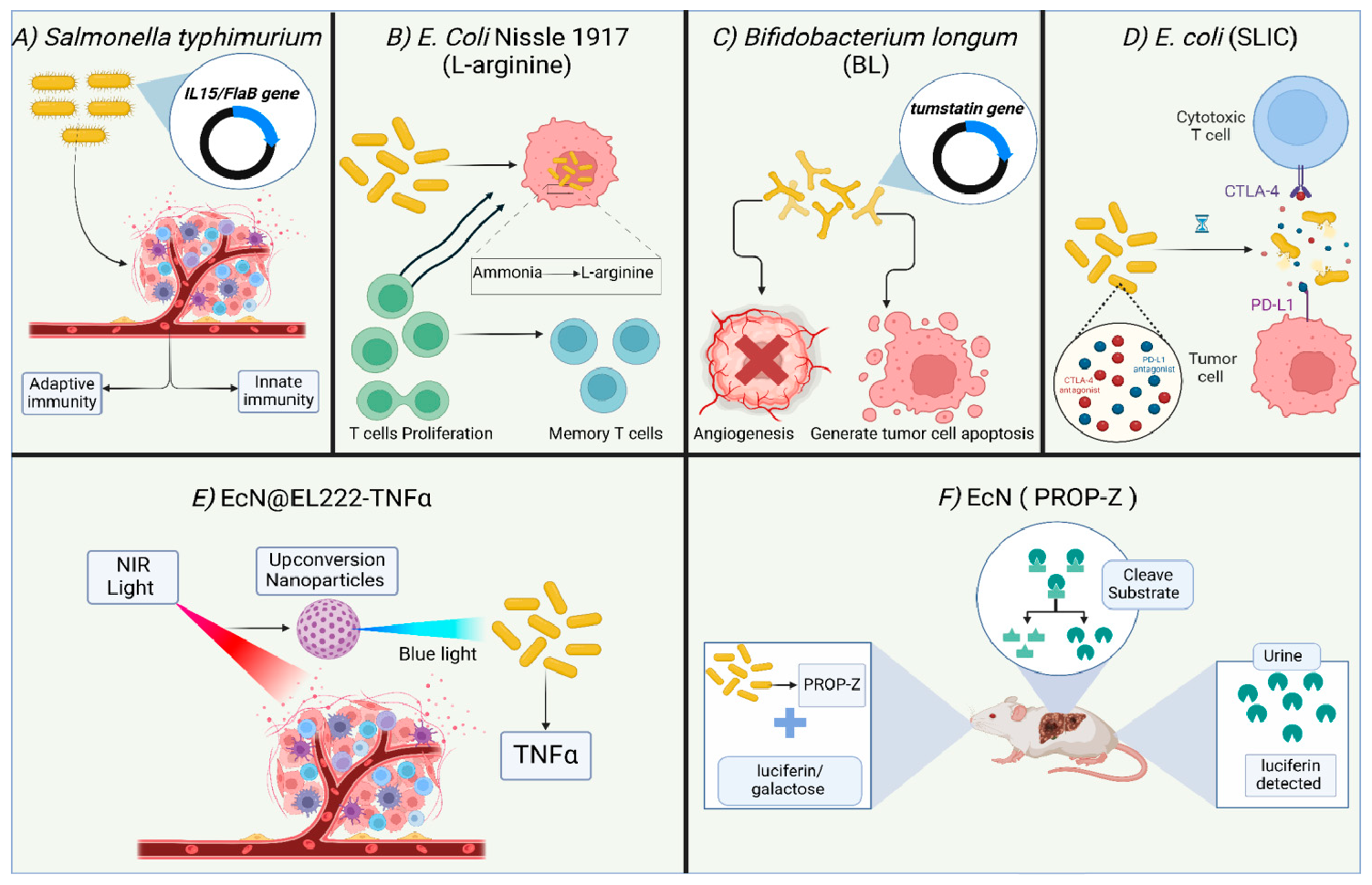
Multiple studies suggest a strong correlation between intratumor microbiota and tumor infiltration of immune cells such as cytotoxic CD8+T cells and Treg cells, exerting either a negative or positive effect on anti-tumor immunity, and implicating tumor progression and clinical outcome [198]. In spite of the paramount significance of intratumor microbiota and its implication in translational application, we still lack a comprehensive understanding of the microbiota-immunity-tumor cells interactions and signals within the TME. Key unanswered questions include how to elucidate the mechanisms underpinning the crosstalk between gut and local tumor microbiota, the impact of intratumor microbes on cancer metastasis, detailed characterization of intratumor microbial communities, large-scale cohorts of clinical studies to determine the impact of intratumor microbes on response to therapeutics and their applications, and the development of microbiome-based diagnostic biomarkers and live therapeutics to enhance activity of cancer therapy. An interesting area of research in this field is the modulation of therapy outcomes through a controlled diet that affects the structure of gut microbes. For example, in mouse models of adenocarcinoma, oral administration of the polysaccharide dietary fiber inulin increased the effectiveness of anti-PD-1 therapy [199]. A trending research in this field is the design and development of engineered probiotics concurrent with the advances and development of next-generation gene editing tools [200,201]. Engineered probiotics could revolutionize cancer diagnosis and treatment protocols, particularly in the targeted delivery of anticancer drugs, provoking anti-tumor immunity, or sensing metastatic tumor cells in a non-invasive manner.
W.K.M. conceptualized the study and review structure, developed tables, figures, and collected and analyzed data. A.A.A. curated data of the microbiome of cancer types, developed figures, and tables. R.W.A.A. collected data related to the application of microbiome therapeutics and managed reference citations. R.A.S. organized and developed tables. N.A.R. collected data on engineered probiotics and developed figures. S.M. collected introductory data on TME. R.G. and TAI developed the review structure and analyzed the literature. All authors wrote, edited, and approved the final version of the manuscript.
This is a review article and does not contain any original data. All sources of information are cited in the reference list.
All authors have read and approved the final manuscript and consent to its publication. There are no conflicts of interest to declare.
The authors declare no conflicts of interest.
This research received no external funding.
Declared none.
[1] Zhan, Y.; Chen, P.-J.; Sadler, W.D.; Wang, F.; Poe, S.; Núñez, G.; Eaton, K.A.; Chen, G.Y. Gut Microbiota Protects against Gastrointestinal Tumorigenesis Caused by Epithelial Injury. Cancer Res. 2013, 73, 7199–7210. [CrossRef]
[2] Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [CrossRef]
[3] Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal Bacteria Control Cancer Response to Therapy by Modulating the Tumor Microenvironment. Science 2013, 342, 967–970. [CrossRef]
[4] Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The Commensal Microbiome Is Associated with anti-PD-1 Efficacy in Metastatic Melanoma Patients. Science 2018, 359, 104–108. [CrossRef]
[5] Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy Against Epithelial Tumors. Science 2018, 359, 91–97. [CrossRef] [PubMed]
[6] Cummins, J.; Tangney, M. Bacteria and Tumours: Causative Agents or Opportunistic Inhabitants?. Infect. Agent. Cancer 2013, 8, 11. [CrossRef]
[7] Baban, C.K.; Cronin, M.; O’Hanlon, D.; O’Sullivan, G.C.; Tangney, M. Bacteria as Vectors for Gene Therapy of Cancer. Bioeng. Bugs 2010, 1, 385–394. [CrossRef] [PubMed]
[8] Gálvez-Cancino, F.; López, E.; Menares, E.; Díaz, X.; Flores, C.; Cáceres, P.; Hidalgo, S.; Chovar, O.; Alcántara-Hernández, M.; Borgna, V.; et al. Vaccination-Induced Skin-Resident Memory CD8+ T Cells Mediate Strong Protection Against Cutaneous Melanoma. Oncoimmunology 2018, 7, e1442163. [CrossRef] [PubMed]
[9] Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of Resident Memory T Cells Enhances the Efficacy of Cancer Vaccine. Nat. Commun. 2017, 8, 15221. [CrossRef]
[10] Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced Anti-Tumour Immunity Requires the Interplay Between Resident and Circulating Memory CD8+ T Cells. Nat. Commun. 2017, 8, 16073. [CrossRef]
[11] Kadoki, M.; Patil, A.; Thaiss, C.C.; Brooks, D.J.; Pandey, S.; Deep, D.; Alvarez, D.; von Andrian, U.H.; Wagers, A.J.; Nakai, K.; et al. Organism-Level Analysis of Vaccination Reveals Networks of Protection Across Tissues. Cell 2017, 171, 398–413.e21. [CrossRef] [PubMed]
[12] Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti–PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [CrossRef]
[13] Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z.; et al. Microbial Translocation Augments the Function of Adoptively Transferred Self/Tumor-Specific CD8+ T Cells via TLR4 Signaling. J. Clin. Invest. 2007, 117, 2197–2204. [CrossRef]
[14] Daillère, R.; Vétizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.M.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus Hirae and Barnesiella Intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 2016, 45, 931–943. [CrossRef] [PubMed]
[15] Rong, Y.; Dong, Z.; Hong, Z.; Jin, Y.; Zhang, W.; Zhang, B.; Mao, W.; Kong, H.; Wang, C.; Yang, B.; et al. Reactivity Toward Bifidobacterium Longum and Enterococcus Hirae Demonstrate Robust CD8+ T Cell Response and Better Prognosis in HBV-Related Hepatocellular Carcinoma. Exp. Cell Res. 2017, 358, 352–359. [CrossRef]
[16] Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [CrossRef] [PubMed]
[17] Qiao, H.; Tan, X.-R.; Li, H.; Li, J.-Y.; Chen, X.-Z.; Li, Y.-Q.; Li, W.-F.; Tang, L.-L.; Zhou, G.-Q.; Zhang, Y.; et al. Association of Intratumoral Microbiota with Prognosis in Patients with Nasopharyngeal Carcinoma From 2 Hospitals in China. JAMA Oncol. 2022, 8, 1301–1309. [CrossRef] [PubMed]
[18] Greathouse, K.L.; White, J.R.; Vargas, A.J.; Bliskovsky, V.V.; Beck, J.A.; von Muhlinen, N.; Polley, E.C.; Bowman, E.D.; Khan, M.A.; Robles, A.I.; et al. Interaction Between the Microbiome and TP53 in Human Lung Cancer. Genome Biol. 2018, 19. [CrossRef]
[19] Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic Microenvironment in Cancer: Molecular Mechanisms and Therapeutic Interventions. Signal Transduct. Target. Ther. 2023, 8, 1–23. [CrossRef]
[20] Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [CrossRef]
[21] Li, T.; Zhao, Z.; Peng, M.; Wang, C.; Luo, F.; Zeng, M.; Sun, K.; Fang, Z.; Luo, Y.; Huang, Q.; et al. Multi-omics Analysis Reveals Novel Interplays Between Intratumoral Bacteria and Glioma. bioRxiv 2023, , 561332.Available online: https://www.biorxiv.org/content/10.1101/2023.10.08.561332v1
[22] Huang, X.; Pan, J.; Xu, F.; Shao, B.; Wang, Y.; Guo, X.; Zhou, S. Bacteria-Based Cancer Immunotherapy. Adv. Sci. 2021, 8, 2003572. [CrossRef] [PubMed]
[23] Hilmi, M.; Kamal, M.; Vacher, S.; Dupain, C.; Ibadioune, S.; Halladjian, M.; Sablin, M.P.; Marret, G.; Ajgal, Z.C.; Nijnikoff, M.; et al. Intratumoral Microbiome Is Driven by Metastatic Site and Associated with Immune Histopathological Parameters: An Ancillary Study of the SHIVA Clinical Trial. Eur. J. Cancer 2023, 183, 152–161. [CrossRef] [PubMed]
[24] Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E.; et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature 2012, 491, 254–258. [CrossRef] [PubMed]
[25] Younginger, B.S.; Mayba, O.; Reeder, J.; Nagarkar, D.R.; Modrusan, Z.; Albert, M.L.; Byrd, A.L. Enrichment of Oral-Derived Bacteria in Inflamed Colorectal Tumors and Distinct Associations of Fusobacterium in the Mesenchymal Subtype. Cell Rep. Med. 2023, 4, 100920. [CrossRef]
[26] Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [CrossRef]
[27] Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Becker, J.; Gonzalez, A.; McKinstry, D.; Hasso, M.; et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [CrossRef]
[28] Li, Y.; Tinoco, R.; Elmén, L.; Segota, I.; Xian, Y.; Fujita, Y.; Sahu, A.; Zarecki, R.; Marie, K.; Feng, Y.; et al. Gut Microbiota Dependent Anti-Tumor Immunity Restricts Melanoma Growth in Rnf5−/− Mice. Nat. Commun. 2019, 10, 1492. [CrossRef]
[29] Mohseni, A.H.; Taghinezhad-S, S.; Keyvani, H. The First Clinical Use of a Recombinant Lactococcus Lactis Expressing Human Papillomavirus Type 16 E7 Oncogene Oral Vaccine: A Phase I Safety and Immunogenicity Trial in Healthy Women Volunteers. Mol. Cancer Ther. 2020, 19, 717–727. [CrossRef]
[30] Taghinezhad-S, S.; Mohseni, A.H.; Keyvani, H.; Razavi, M.R. Phase 1 Safety and Immunogenicity Trial of Recombinant Lactococcus Lactis Expressing Human Papillomavirus Type 16 E6 Oncoprotein Vaccine. Mol. Ther. Methods Clin. Dev. 2019, 15, 40–51. [CrossRef]
[31] Mohseni, A.H.; Razavilar, V.; Keyvani, H.; Razavi, M.R.; Khavari-Nejad, R.A. Oral Immunization with Recombinant Lactococcus Lactis NZ9000 Expressing Human Papillomavirus Type 16 E7 Antigen and Evaluation of Its Immune Effects in Female C57BL/6 Mice. J. Med. Virol. 2019, 91, 296–307. [CrossRef]
[32] Wu, Y.; Kyle-Cezar, F.; Woolf, R.T.; Naceur-Lombardelli, C.; Owen, J.; Biswas, D.; Lorenc, A.; Vantourout, P.; Gazinska, P.; Grigoriadis, A.; et al. An Innate-Like Vδ1+ γδ T Cell Compartment in the Human Breast Is Associated with Remission in Triple-Negative Breast Cancer. Sci. Transl. Med. 2019, 11, eaax9364. [CrossRef]
[33] Zegarra-Ruiz, D.F.; Kim, D.V.; Norwood, K.; Kim, M.; Wu, W.-J.H.; Saldana-Morales, F.B.; Hill, A.A.; Majumdar, S.; Orozco, S.; Bell, R.; et al. Thymic Development of Gut-Microbiota-Specific T Cells. Nature 2021, 594, 413–417. [CrossRef]
[34] Cebula, A.; Seweryn, M.; Rempala, G.A.; Pabla, S.S.; McIndoe, R.A.; Denning, T.L.; Bry, L.; Kraj, P.; Kisielow, P.; Ignatowicz, L. Thymus-Derived Regulatory T Cells Contribute to Tolerance to Commensal Microbiota. Nature 2013, 497, 258–262. [CrossRef] [PubMed]
[35] Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut Microbiome-Mediated Bile Acid Metabolism Regulates Liver Cancer via NKT Cells. Science 2018, 360, eaan5931. [CrossRef]
[36] Guo, W.; Zhang, Y.; Guo, S.; Mei, Z.; Liao, H.; Dong, H.; Wu, K.; Ye, H.; Zhang, Y.; Zhu, Y.; et al. Tumor Microbiome Contributes to an Aggressive Phenotype in the Basal-Like Subtype of Pancreatic Cancer. Commun. Biol. 2021, 4. [CrossRef] [PubMed]
[37] Peeters, P.J.H.L.; Bazelier, M.T.; Leufkens, H.G.M.; de Vries, F.; De Bruin, M.L. The Risk of Colorectal Cancer in Patients with Type 2 Diabetes: Associations with Treatment Stage and Obesity. Diabetes Care 2015, 38, 495–502. [CrossRef] [PubMed]
[38] Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [CrossRef]
[39] Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow?. The Lancet 2001, 357, 539–545. [CrossRef]
[40] Tsay, J.-C.J.; Wu, B.G.; Sulaiman, I.; Gershner, K.; Schluger, R.; Li, Y.; Yie, T.-A.; Meyn, P.; Olsen, E.; Perez, L.; et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov. 2021, 11, 293–307. [CrossRef]
[41] Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e16. [CrossRef]
[42] Triner, D.; Devenport, S.N.; Ramakrishnan, S.K.; Ma, X.; Frieler, R.A.; Greenson, J.K.; Inohara, N.; Nunez, G.; Colacino, J.A.; Mortensen, R.M.; et al. Neutrophils Restrict Tumor-Associated Microbiota to Reduce Growth and Invasion of Colon Tumors in Mice. Gastroenterology 2019, 156, 1467–1482. [CrossRef] [PubMed]
[43] Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate Sensing of Microbial Products Promotes Wound-Induced Skin Cancer. Nat. Commun. 2015, 6, 5932. [CrossRef] [PubMed]
[44] Hayashi, M.; Ikenaga, N.; Nakata, K.; Luo, H.; Zhong, P.; Date, S.; Oyama, K.; Higashijima, N.; Kubo, A.; Iwamoto, C.; et al. Intratumor Fusobacterium Nucleatum Promotes the Progression of Pancreatic Cancer via the CXCL1-CXCR2 Axis. Cancer Sci. 2023, 114, 3666–3678. [CrossRef] [PubMed]
[45] Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.P.; Namini, M.R.J.; Hansen, M.B.; Martin, O.C.B.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial Genotoxins Induce T Cell Senescence. Cell Rep. 2021, 35. [CrossRef]
[46] Halley, A.; Leonetti, A.; Gregori, A.; Tiseo, M.; Deng, D.M.; Giovannetti, E.; Peters, G.J. The Role of the Microbiome in Cancer and Therapy Efficacy: Focus on Lung Cancer. Anticancer Res. 2020, 40, 4807–4818. [CrossRef]
[47] Balmer, M.L.; Ma, E.H.; Bantug, G.R.; Grählert, J.; Pfister, S.; Glatter, T.; Jauch, A.; Dimeloe, S.; Slack, E.; Dehio, P.; et al. Memory CD8(+) T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 2016, 44, 1312–1324. [CrossRef]
[48] Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c− Patrolling Monocyte Hematopoiesis and CD8+ T Cell Metabolism. Immunity 2018, 48, 992–1005.e8. [CrossRef]
[49] Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the Effector Function of CD8+ T Cells by Gut Microbiota-Derived Metabolite Butyrate. Sci. Rep. 2018, 8. [CrossRef]
[50] Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary Fiber and Probiotics Influence the Gut Microbiome and Melanoma Immunotherapy Response. Science 2021, 374, 1632–1640. [CrossRef]
[51] Broadfield, L.A.; Saigal, A.; Szamosi, J.C.; Hammill, J.A.; Bezverbnaya, K.; Wang, D.; Gautam, J.; Tsakiridis, E.E.; Di Pastena, F.; McNicol, J.; et al. Metformin-Induced Reductions in Tumor Growth Involves Modulation of the Gut Microbiome. Mol. Metab. 2022, 61, 101498. [CrossRef]
[52] Zhu, G.; Su, H.; Johnson, C.H.; Khan, S.A.; Kluger, H.; Lu, L. Intratumour Microbiome Associated with the Infiltration of Cytotoxic CD8+ T Cells and Patient Survival in Cutaneous Melanoma. Eur. J. Cancer 2021, 151, 25–34. [CrossRef]
[53] Barrett, M.; Hand, C.K.; Shanahan, F.; Murphy, T.; O’Toole, P.W. Mutagenesis by Microbe: The Role of the Microbiota in Shaping the Cancer Genome. Trends Cancer 2020, 6, 277–287. [CrossRef]
[54] Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The Microbiome and Human Cancer. Science 2021, 371, eabc4552. [CrossRef] [PubMed]
[55] Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks+ E. coli. Nature 2020, 580, 269–273. [CrossRef] [PubMed]
[56] Allen, J.; Rosendahl Huber, A.; Pleguezuelos-Manzano, C.; Puschhof, J.; Wu, S.; Wu, X.; Boot, C.; Saftien, A.; O’Hagan, H.M.; Wang, H.; et al. Colon Tumors in Enterotoxigenic Bacteroides fragilis (ETBF)-Colonized Mice Do Not Display a Unique Mutational Signature but Instead Possess Host-Dependent Alterations in the APC Gene. Microbiol. Spectr. 2022, 10, e01055-22. [CrossRef] [PubMed]
[57] Cheng, W.T.; Kantilal, H.K.; Davamani, F. The Mechanism of Bacteroides fragilis Toxin Contributes to Colon Cancer Formation. Malays. J. Med. Sci. MJMS 2020, 27, 9. [CrossRef]
[58] Goodwin, A.C.; Shields, C.E.D.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine Catabolism Contributes to Enterotoxigenic Bacteroides Fragilis-Induced Colon Tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [CrossRef]
[59] Cao, Y.; Oh, J.; Xue, M.; Huh, W.J.; Wang, J.; Gonzalez-Hernandez, J.A.; Rice, T.A.; Martin, A.L.; Song, D.; Crawford, J.M.; et al. Commensal Microbiota from Patients with Inflammatory Bowel Disease Produce Genotoxic Metabolites. Science 2022, 378, eabm3233. [CrossRef]
[60] Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annu. Rev. Biochem. 2005, 74, 681–710. [CrossRef]
[61] Bateman, A.C. DNA Mismatch Repair Proteins: Scientific Update and Practical Guide. J. Clin. Pathol. 2021, 74, 264–268. [CrossRef]
[62] Santos, J.C.; Brianti, M.T.; Almeida, V.R.; Ortega, M.M.; Fischer, W.; Haas, R.; Matheu, A.; Ribeiro, M.L. Helicobacter Pylori Infection Modulates the Expression of miRNAs Associated with DNA Mismatch Repair Pathway. Mol. Carcinog. 2017, 56, 1372–1379. [CrossRef]
[63] Abreu, M.T.; Peek, R.M. Gastrointestinal Malignancy and the Microbiome. Gastroenterology 2014, 146, 1534–1546.e3. [CrossRef] [PubMed]
[64] Banerjee, S.; Alwine, J.C.; Wei, Z.; Tian, T.; Shih, N.; Sperling, C.; Guzzo, T.; Feldman, M.D.; Robertson, E.S. Microbiome Signatures in Prostate Cancer. Carcinogenesis 2019, 40, 749–764. [CrossRef]
[65] Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of β-Catenin by Carcinogenic Helicobacter Pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [CrossRef] [PubMed]
[66] Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [CrossRef]
[67] Dadgar-Zankbar, L.; Shariati, A.; Bostanghadiri, N.; Elahi, Z.; Mirkalantari, S.; Razavi, S.; Kamali, F.; Darban-Sarokhalil, D. Evaluation of Enterotoxigenic Bacteroides Fragilis Correlation with the Expression of Cellular Signaling Pathway Genes in Iranian Patients with Colorectal Cancer. Infect. Agent. Cancer 2023, 18, 48. [CrossRef]
[68] Lu, R.; Wu, S.; Zhang, Y.-g.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric Bacterial Protein AvrA Promotes Colonic Tumorigenesis and Activates Colonic Beta-Catenin Signaling Pathway. Oncogenesis 2014, 3, e105. [CrossRef] [PubMed]
[69] Peng, R.; Liu, S.; You, W.; Huang, Y.; Hu, C.; Gao, Y.; Jia, X.; Li, G.; Xu, Z.; Chen, Y. Gastric Microbiome Alterations Are Associated with Decreased CD8+ Tissue-Resident Memory T Cells in the Tumor Microenvironment of Gastric Cancer. Cancer Immunol. Res. 2022, 10, 1224–1240. [CrossRef]
[70] Zhang, J.; Zhang, F.; Zhao, C.; Xu, Q.; Liang, C.; Yang, Y.; Wang, H.; Shang, Y.; Wang, Y.; Mu, X.; et al. Dysbiosis of the Gut Microbiome Is Associated with Thyroid Cancer and Thyroid Nodules and Correlated with Clinical Index of Thyroid Function. Endocrine 2019, 64, 564–574. [CrossRef]
[71] Yuan, L.; Yang, P.; Wei, G.; Hu, X.; Chen, S.; Lu, J.; Yang, L.; He, X.; Bao, G. Tumor Microbiome Diversity Influences Papillary Thyroid Cancer Invasion. Commun. Biol. 2022, 5. [CrossRef]
[72] Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and Laboratory Contamination Can Critically Impact Sequence-Based Microbiome Analyses. BMC Biol. 2014, 12. [CrossRef] [PubMed]
[73] Hogan, G.; Eckenberger, J.; Narayanen, N.; Walker, S.P.; Claesson, M.J.; Corrigan, M.; O’Hanlon, D.; Tangney, M. Biopsy Bacterial Signature Can Predict Patient Tissue Malignancy. Sci. Rep. 2021, 11. [CrossRef]
[74] Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [CrossRef] [PubMed]
[75] Czarnecka-Chrebelska, K.H.; Kordiak, J.; Brzeziańska-Lasota, E.; Pastuszak-Lewandoska, D. Respiratory Tract Oncobiome in Lung Carcinogenesis: Where Are We Now?. Cancers 2023, 15. [CrossRef] [PubMed]
[76] Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The Human Tumor Microbiome Is Composed of Tumor Type-Specific Intracellular Bacteria. Science 2020, 368, 973–980. [CrossRef]
[77] Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806.e12. [CrossRef]
[78] Chang, Y.-S.; Hsu, M.-H.; Tu, S.-J.; Yen, J.-C.; Lee, Y.-T.; Fang, H.-Y.; Chang, J.-G. Metatranscriptomic Analysis of Human Lung Metagenomes from Patients with Lung Cancer. Genes 2021, 12. [CrossRef]
[79] Boesch, M.; Baty, F.; Albrich, W.C.; Flatz, L.; Rodriguez, R.; Rothschild, S.I.; Joerger, M.; Früh, M.; Brutsche, M.H. Local Tumor Microbial Signatures and Response to Checkpoint Blockade in Non-Small Cell Lung Cancer. Oncoimmunology 2021, 10, 1988403. [CrossRef]
[80] Tzeng, A.; Sangwan, N.; Jia, M.; Liu, C.-C.; Keslar, K.S.; Downs-Kelly, E.; Fairchild, R.L.; Al-Hilli, Z.; Grobmyer, S.R.; Eng, C. Human Breast Microbiome Correlates with Prognostic Features and Immunological Signatures in Breast Cancer. Genome Med. 2021, 13, 60. [CrossRef]
[81] Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 2016, 6. [CrossRef]
[82] Qu, D.; Wang, Y.; Xia, Q.; Chang, J.; Jiang, X.; Zhang, H. Intratumoral Microbiome of Human Primary Liver Cancer. Hepatol. Commun. 2022, 6, 1741. [CrossRef] [PubMed]
[83] Sun, L.; Ke, X.; Guan, A.; Jin, B.; Qu, J.; Wang, Y.; Xu, X.; Li, C.; Sun, H.; Xu, H.; et al. Intratumoural Microbiome Can Predict the Prognosis of Hepatocellular Carcinoma After Surgery. Clin. Transl. Med. 2023, 13, e1331. [CrossRef]
[84] Jiang, L.; Duan, B.; Jia, P.; Zhang, Y.; Yan, X. The Role of Intratumor Microbiomes in Cervical Cancer Metastasis. Cancers 2023, 15. [CrossRef] [PubMed]
[85] Liu, Z.; Zhang, X.; Zhang, H.; Zhang, H.; Yi, Z.; Zhang, Q.; Liu, Q.; Liu, X. Multi-Omics Analysis Reveals Intratumor Microbes as Immunomodulators in Colorectal Cancer. Microbiol. Spectr. 2023, 11, e05038-22. [CrossRef] [PubMed]
[86] Kullander, J.; Forslund, O.; Dillner, J. Staphylococcus aureus and Squamous Cell Carcinoma of the Skin. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 472–478. [CrossRef]
[87] Wang, J.; Li, X.; Wu, X.; Wang, Z.; Zhang, C.; Cao, G.; Liu, K.; Yan, T. Uncovering the Microbiota in Renal Cell Carcinoma Tissue Using 16S rRNA Gene Sequencing. J. Cancer Res. Clin. Oncol. 2021, 147, 481–491. [CrossRef]
[88] Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Lucianò, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The Microbiome of the Prostate Tumor Microenvironment. Eur. Urol. 2017, 72, 625–631. [CrossRef]
[89] Li, W.T.; Iyangar, A.S.; Reddy, R.; Chakladar, J.; Bhargava, V.; Sakamoto, K.; Ongkeko, W.M.; Rajasekaran, M. The Bladder Microbiome Is Associated with Epithelial–Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma. Cancers 2021, 13. [CrossRef]
[90] Zhou, B.; Sun, C.; Huang, J.; Xia, M.; Guo, E.; Li, N.; Lu, H.; Shan, W.; Wu, Y.; Li, Y.; et al. The Biodiversity Composition of Microbiome in Ovarian Carcinoma Patients. Sci. Rep. 2019, 9. [CrossRef]
[91] Hill, M.J.; Goddard, P.; Williams, R.E.O. Gut Bacteria and Ætiology of Cancer of the Breast. Lancet 1971, 298, 472–473. [CrossRef]
[92] Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [CrossRef] [PubMed]
[93] Meng, S.; Chen, B.; Yang, J.; Wang, J.; Zhu, D.; Meng, Q.; Zhang, L. Study of Microbiomes in Aseptically Collected Samples of Human Breast Tissue Using Needle Biopsy and the Potential Role of in situ Tissue Microbiomes for Promoting Malignancy. Front. Oncol. 2018, 8. [CrossRef] [PubMed]
[94] Banerjee, S.; Wei, Z.; Tian, T.; Bose, D.; Shih, N.N.C.; Feldman, M.D.; Khoury, T.; De Michele, A.; Robertson, E.S. Prognostic Correlations with the Microbiome of Breast Cancer Subtypes. Cell Death Dis. 2021, 12, 1–14. [CrossRef]
[95] del Castillo, E.; Meier, R.; Chung, M.; Koestler, D.C.; Chen, T.; Paster, B.J.; Charpentier, K.P.; Kelsey, K.T.; Izard, J.; Michaud, D.S. The Microbiomes of Pancreatic and Duodenum Tissue Overlap and Are Highly Subject Specific but Differ Between Pancreatic Cancer and Noncancer Subjects. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 370–383. [CrossRef]
[96] Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential Role of Intratumor Bacteria in Mediating Tumor Resistance to the Chemotherapeutic Drug Gemcitabine. Science 2017, 357, 1156–1160. [CrossRef]
[97] Öğrendik, M. Periodontal Pathogens in the Etiology of Pancreatic Cancer. Gastrointest. Tumors 2016, 3, 125–127. [CrossRef]
[98] Tan, Q.; Ma, X.; Yang, B.; Liu, Y.; Xie, Y.; Wang, X.; Yuan, W.; Ma, J. Periodontitis Pathogen Porphyromonas Gingivalis Promotes Pancreatic Tumorigenesis via Neutrophil Elastase from Tumor-Associated Neutrophils. Gut Microbes 2022, 14, 2073785. [CrossRef] [PubMed]
[99] Js, C.; Cr, T.; Lt, C.; Ys, S. Investigating the Association Between Periodontal Disease and Risk of Pancreatic Cancer. Pancreas 2016, 45. [CrossRef]
[100] Ungureanu, B.S.; Gheorghe, D.N.; Nicolae, F.M.; Râmboiu, S.; Radu, P.A.; Șurlin, V.M.; Strâmbu, V.D.E.; Gheonea, D.I.; Roman, A.; Șurlin, P. Could There Be an Interplay Between Periodontal Changes and Pancreatic Malignancies?. World J. Clin. Cases 2023, 11, 545–555. [CrossRef]
[101] Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium Species in Pancreatic Cancer Tissues with Molecular Features and Prognosis. Oncotarget 2015, 6, 7209–7220. [CrossRef]
[102] Chakladar, J.; Kuo, S.Z.; Castaneda, G.; Li, W.T.; Gnanasekar, A.; Yu, M.A.; Chang, E.Y.; Wang, X.Q.; Ongkeko, W.M. The Pancreatic Microbiome Is Associated with Carcinogenesis and Worse Prognosis in Males and Smokers. Cancers 2020, 12. [CrossRef] [PubMed]
[103] Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA. Cancer J. Clin. 2020, 70, 145–164. [CrossRef] [PubMed]
[104] da Costa, C.P.; Vieira, P.; Mendes-Rocha, M.; Pereira-Marques, J.; Ferreira, R.M.; Figueiredo, C. The Tissue-Associated Microbiota in Colorectal Cancer: A Systematic Review. Cancers 2022, 14. [CrossRef] [PubMed]
[105] Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [CrossRef]
[106] Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum Infection Is Prevalent in Human Colorectal Carcinoma. Genome Res. 2012, 22, 299–306. [CrossRef]
[107] Yamamoto, S.; Kinugasa, H.; Hirai, M.; Terasawa, H.; Yasutomi, E.; Oka, S.; Ohmori, M.; Yamasaki, Y.; Inokuchi, T.; Harada, K.; et al. Heterogeneous Distribution of Fusobacterium Nucleatum in the Progression of Colorectal Cancer. J. Gastroenterol. Hepatol. 2021, 36, 1869–1876. [CrossRef]
[108] Hamada, T.; Zhang, X.; Mima, K.; Bullman, S.; Sukawa, Y.; Nowak, J.A.; Kosumi, K.; Masugi, Y.; Twombly, T.S.; Cao, Y.; et al. Fusobacterium Nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability Status. Cancer Immunol. Res. 2018, 6, 1327–1336. [CrossRef]
[109] Zhang, Y.; Zhang, L.; Zheng, S.; Li, M.; Xu, C.; Jia, D.; Qi, Y.; Hou, T.; Wang, L.; Wang, B.; et al. Fusobacterium Nucleatum Promotes Colorectal Cancer Cells Adhesion to Endothelial Cells and Facilitates Extravasation and Metastasis by Inducing ALPK1/NF-κB/ICAM1 Axis. Gut Microbes 2022, 14, 2038852. [CrossRef]
[110] Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [CrossRef]
[111] Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium Nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via TLR4/Keap1/NRF2 Signaling. Cancer Res. 2021, 81, 4485–4498. [CrossRef]
[112] Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e24. [CrossRef] [PubMed]
[113] Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of Mucosa-Associated E. coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS ONE 2013, 8. [CrossRef] [PubMed]
[114] He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter Jejuni Promotes Colorectal Tumorigenesis Through the Action of Cytolethal Distending Toxin. Gut 2019, 68, 289–300. [CrossRef]
[115] Okuda, S.; Shimada, Y.; Tajima, Y.; Yuza, K.; Hirose, Y.; Ichikawa, H.; Nagahashi, M.; Sakata, J.; Ling, Y.; Miura, N.; et al. Profiling of Host Genetic Alterations and Intra-Tumor Microbiomes in Colorectal Cancer. Comput. Struct. Biotechnol. J. 2021, 19, 3330–3338. [CrossRef]
[116] Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-Occurrence of Anaerobic Bacteria in Colorectal Carcinomas. Microbiome 2013, 1. [CrossRef] [PubMed]
[117] Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the Human Colorectal Cancer Microbiome. PLOS ONE 2011, 6. [CrossRef]
[118] Thrift, A.P.; Wenker, T.N.; El-Serag, H.B. Global Burden of Gastric Cancer: Epidemiological Trends, Risk Factors, Screening and Prevention. Nat. Rev. Clin. Oncol. 2023, 20, 338–349. [CrossRef]
[119] Li, Q.; Wu, W.; Gong, D.; Shang, R.; Wang, J.; Yu, H. Propionibacterium Acnes Overabundance in Gastric Cancer Promote M2 Polarization of Macrophages via a TLR4/PI3K/Akt Signaling. Gastric Cancer 2021, 24, 1242–1253. [CrossRef]
[120] de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob. Health 2020, 8, e180–e190. [CrossRef]
[121] Helicobacter and Cancer Collaborative Group Gastric Cancer and Helicobacter Pylori: A Combined Analysis of 12 Case Control Studies Nested Within Prospective Cohorts. Gut 2001, 49, 347–353. [CrossRef]
[122] Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of Gastric Mucosal Microbiota Across Different Stomach Microhabitats in a Cohort of 276 Patients with Gastric Cancer. eBioMedicine 2019, 40, 336–348. [CrossRef] [PubMed]
[123] Liou, J.-M.; Malfertheiner, P.; Lee, Y.-C.; Sheu, B.-S.; Sugano, K.; Cheng, H.-C.; Yeoh, K.-G.; Hsu, P.-I.; Goh, K.-L.; Mahachai, V.; et al. Screening and Eradication of Helicobacter Pylori for Gastric Cancer Prevention: The Taipei Global Consensus. Gut 2020, 69, 2093–2112. [CrossRef]
[124] Yu, G.; Torres, J.; Hu, N.; Medrano-Guzman, R.; Herrera-Goepfert, R.; Humphrys, M.S.; Wang, L.; Wang, C.; Ding, T.; Ravel, J.; et al. Molecular Characterization of the Human Stomach Microbiota in Gastric Cancer Patients. Front. Cell. Infect. Microbiol. 2017, 7. [CrossRef] [PubMed]
[125] Noto, J.M.; Zackular, J.P.; Varga, M.G.; Delgado, A.; Romero-Gallo, J.; Scholz, M.B.; Piazuelo, M.B.; Skaar, E.P.; Peek, R.M. Modification of the Gastric Mucosal Microbiota by a Strain-Specific Helicobacter Pylori Oncoprotein and Carcinogenic Histologic Phenotype. mBio 2019, 10, 10.1128/mbio.00955-19. [CrossRef] [PubMed]
[126] Apostolou, P.; Tsantsaridou, A.; Papasotiriou, I.; Toloudi, M.; Chatziioannou, M.; Giamouzis, G. Bacterial and Fungal Microflora in Surgically Removed Lung Cancer Samples. J. Cardiothorac. Surg. 2011, 6. [CrossRef]
[127] Lee, S.H.; Sung, J.Y.; Yong, D.; Chun, J.; Kim, S.Y.; Song, J.H.; Chung, K.S.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; et al. Characterization of Microbiome in Bronchoalveolar Lavage Fluid of Patients with Lung Cancer Comparing with Benign Mass Like Lesions. Lung Cancer 2016, 102, 89–95. [CrossRef]
[128] Druzhinin, V.G.; Matskova, L.V.; Demenkov, P.S.; Baranova, E.D.; Volobaev, V.P.; Minina, V.I.; Larionov, A.V.; Titov, V.A.; Fucic, A. Genetic Damage in Lymphocytes of Lung Cancer Patients Is Correlated to the Composition of the Respiratory Tract Microbiome. Mutagenesis 2021, 36, 143–153. [CrossRef]
[129] Liu, H.-X.; Tao, L.-L.; Zhang, J.; Zhu, Y.-G.; Zheng, Y.; Liu, D.; Zhou, M.; Ke, H.; Shi, M.-M.; Qu, J.-M. Difference of Lower Airway Microbiome in Bilateral Protected Specimen Brush Between Lung Cancer Patients with Unilateral Lobar Masses and Control Subjects. Int. J. Cancer 2018, 142, 769–778. [CrossRef]
[130] Bingula, R.; Filaire, E.; Molnar, I.; Delmas, E.; Berthon, J.-Y.; Vasson, M.-P.; Bernalier-Donadille, A.; Filaire, M. Characterisation of Microbiota in Saliva, Bronchoalveolar Lavage Fluid, Non-Malignant, Peritumoural and Tumour Tissue in Non-Small Cell Lung Cancer Patients: A Cross-Sectional Clinical Trial. Respir. Res. 2020, 21, 129. [CrossRef]
[131] Patnaik, S.K.; Cortes, E.G.; Kannisto, E.D.; Punnanitinont, A.; Dhillon, S.S.; Liu, S.; Yendamuri, S. Lower Airway Bacterial Microbiome May Influence Recurrence After Resection of Early-Stage Non–Small Cell Lung Cancer. J. Thorac. Cardiovasc. Surg. 2021, 161, 419–429.e16. [CrossRef]
[132] Zhang, M.; Zhang, Y.; Sun, Y.; Wang, S.; Liang, H.; Han, Y. Intratumoral Microbiota Impacts the First-Line Treatment Efficacy and Survival in Non-Small Cell Lung Cancer Patients Free of Lung Infection. J. Healthc. Eng. 2022, 2022, 5466853. [CrossRef] [PubMed]
[133] Yuan, X.; Wang, Z.; Li, C.; Lv, K.; Tian, G.; Tang, M.; Ji, L.; Yang, J. Bacterial Biomarkers Capable of Identifying Recurrence or Metastasis Carry Disease Severity Information for Lung Cancer. Front. Microbiol. 2022, 13. [CrossRef] [PubMed]
[134] Oh, K.; By, C.; Dk, K.; Nh, K.; Jk, R.; Wj, S.; Sw, L. The Microbiome of Lung Cancer Tissue and Its Association with Pathological and Clinical Parameters. Am. J. Cancer Res. 2022, 12, 2350–2362.https://pmc.ncbi.nlm.nih.gov/articles/PMC9185621/
[135] Apopa, P.L.; Alley, L.; Penney, R.B.; Arnaoutakis, K.; Steliga, M.A.; Jeffus, S.; Bircan, E.; Gopalan, B.; Jin, J.; Patumcharoenpol, P.; et al. PARP1 Is Up-Regulated in Non-Small Cell Lung Cancer Tissues in the Presence of the Cyanobacterial Toxin Microcystin. Front. Microbiol. 2018, 9. [CrossRef] [PubMed]
[136] Gomes, S.; Cavadas, B.; Ferreira, J.C.; Marques, P.I.; Monteiro, C.; Sucena, M.; Sousa, C.; Vaz Rodrigues, L.; Teixeira, G.; Pinto, P.; et al. Profiling of Lung Microbiota Discloses Differences in Adenocarcinoma and Squamous Cell Carcinoma. Sci. Rep. 2019, 9. [CrossRef]
[137] Wong, L.M.; Shende, N.; Li, W.T.; Castaneda, G.; Apostol, L.; Chang, E.Y.; Ongkeko, W.M. Comparative Analysis of Age- and Gender-Associated Microbiome in Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. Cancers 2020, 12. [CrossRef]
[138] Dong, H.; Tan, Q.; Xu, Y.; Zhu, Y.; Yao, Y.; Wang, Y.; Li, C.; Li, H.; Zhang, G.; Xiong, Y.; et al. Convergent Alteration of Lung Tissue Microbiota and Tumor Cells in Lung Cancer. iScience 2022, 25. [CrossRef]
[139] Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global Burden of Primary Liver Cancer in 2020 and Predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [CrossRef]
[140] Kang, Y.; Cai, Y.; Yang, Y. The Gut Microbiome and Hepatocellular Carcinoma: Implications for Early Diagnostic Biomarkers and Novel Therapies. Liver Cancer 2021, 11, 113–125. [CrossRef]
[141] Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [CrossRef]
[142] Enterococcus Faecalis Colonization in the Gut Promotes Liver Carcinogenesis. Cancer Discov. 2021, 11, 2955. [CrossRef] [PubMed]
[143] Sandri, G.B.L.; Ettorre, G.M.; Colasanti, M.; Werra, E.D.; Mascian? , G.; Ferraro, D.; Tortorelli, G.; Sciuto, R.; Lucatelli, P.; Pizzi, G.; et al. Hepatocellular Carcinoma with Macrovascular Invasion Treated with Yttrium-90 Radioembolization Prior to Transplantation. Hepatobiliary Surg. Nutr. 2017, 6, 448. [CrossRef] [PubMed]
[144] Song, Y.; Xiang, Z.; Lu, Z.; Su, R.; Shu, W.; Sui, M.; Wei, X.; Xu, X. Identification of a Brand Intratumor Microbiome Signature for Predicting Prognosis of Hepatocellular Carcinoma. J. Cancer Res. Clin. Oncol. 2023, 149, 11319–11332. [CrossRef] [PubMed]
[145] Abedi, E.; Hashemi, S.M.B. Lactic Acid Production—Producing Microorganisms and Substrates Sources-State of Art. Heliyon 2020, 6. [CrossRef]
[146] Colbert, L.E.; Alam, M.B.E.; Wang, R.; Karpinets, T.; Lo, D.; Lynn, E.J.; Harris, T.A.; Elnaggar, J.H.; Yoshida-Court, K.; Tomasic, K.; et al. Tumor-Resident Lactobacillus Iners Confer Chemoradiation Resistance Through Lactate-Induced Metabolic Rewiring. Cancer Cell 2023, 41, 1945–1962.e11. [CrossRef]
[147] Grice, E.A.; Kong, H.H.; Renaud, G.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Wolfsberg, T.G.; Turner, M.L.; Segre, J.A. A Diversity Profile of the Human Skin Microbiota. Genome Res. 2008, 18, 1043–1050. [CrossRef]
[148] Byrd, A.L.; Belkaid, Y.; Segre, J.A. The Human Skin Microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [CrossRef]
[149] Madhusudhan, N.; Pausan, M.R.; Halwachs, B.; Durdević, M.; Windisch, M.; Kehrmann, J.; Patra, V.; Wolf, P.; Boukamp, P.; Moissl-Eichinger, C.; et al. Molecular Profiling of Keratinocyte Skin Tumors Links Staphylococcus Aureus Overabundance and Increased Human β-Defensin-2 Expression to Growth Promotion of Squamous Cell Carcinoma. Cancers 2020, 12. [CrossRef]
[150] Hosen, M.E.; Jahan Supti, S.; Akash, S.; Rahman, M.E.; Faruqe, M.O.; Manirujjaman, M.; Acharjee, U.K.; Gaafar, A.-R.Z.; Ouahmane, L.; Sitotaw, B.; et al. Mechanistic Insight of Staphylococcus Aureus Associated Skin Cancer in Humans by Santalum Album Derived Phytochemicals: An Extensive Computational and Experimental Approaches. Front. Chem. 2023, 11. [CrossRef]
[151] Nakatsuji, T.; Chen, T.H.; Butcher, A.M.; Trzoss, L.L.; Nam, S.-J.; Shirakawa, K.T.; Zhou, W.; Oh, J.; Otto, M.; Fenical, W.; et al. A Commensal Strain of Staphylococcus Epidermidis Protects Against Skin Neoplasia. Sci. Adv. 2018, 4, eaao4502. [CrossRef]
[152] Tsuda, K.; Yamanaka, K.; Linan, W.; Miyahara, Y.; Akeda, T.; Nakanishi, T.; Kitagawa, H.; Kakeda, M.; Kurokawa, I.; Shiku, H.; et al. Intratumoral Injection of Propionibacterium Acnes Suppresses Malignant Melanoma by Enhancing Th1 Immune Responses. PLoS ONE 2011, 6. [CrossRef] [PubMed]
[153] Ma, J.; Gnanasekar, A.; Lee, A.; Li, W.T.; Haas, M.; Wang-Rodriguez, J.; Chang, E.Y.; Rajasekaran, M.; Ongkeko, W.M. Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer. Cancers 2020, 12. [CrossRef] [PubMed]
[154] Fassi Fehri, L.; Mak, T.N.; Laube, B.; Brinkmann, V.; Ogilvie, L.A.; Mollenkopf, H.; Lein, M.; Schmidt, T.; Meyer, T.F.; Brüggemann, H. Prevalence of Propionibacterium acnes in Diseased Prostates and Its Inflammatory and Transforming Activity on Prostate Epithelial Cells. Int. J. Med. Microbiol. 2011, 301, 69–78. [CrossRef]
[155] Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Coukos, G.; Alwine, J.C.; Robertson, E.S. The Ovarian Cancer Oncobiome. Oncotarget 2017, 8, 36225–36245. [CrossRef] [PubMed]
[156] Mansour, B.; Monyók, Á.; Makra, N.; Gajdács, M.; Vadnay, I.; Ligeti, B.; Juhász, J.; Szabó, D.; Ostorházi, E. Bladder Cancer-Related Microbiota: Examining Differences in Urine and Tissue Samples. Sci. Rep. 2020, 10, 1–10. [CrossRef]
[157] Bender, M.J.; McPherson, A.C.; Phelps, C.M.; Pandey, S.P.; Laughlin, C.R.; Shapira, J.H.; Sanchez, L.M.; Rana, M.; Richie, T.G.; Mims, T.S.; et al. Dietary Tryptophan Metabolite Released by Intratumoral Lactobacillus Reuteri Facilitates Immune Checkpoint Inhibitor Treatment. Cell 2023, 186, 1846–1862.e26. [CrossRef]
[158] Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A Defined Commensal Consortium Elicits CD8 T Cells and Anti-Cancer Immunity. Nature 2019, 565, 600–605. [CrossRef]
[159] Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal Microbiota Transplant Overcomes Resistance to anti–PD-1 Therapy in Melanoma Patients. Science 2021, 371, 595–602. [CrossRef]
[160] Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science 2021, 371, 602–609. [CrossRef]
[161] Smith, M.; Dai, A.; Ghilardi, G.; Amelsberg, K.V.; Devlin, S.M.; Pajarillo, R.; Slingerland, J.B.; Beghi, S.; Herrera, P.S.; Giardina, P.; et al. Gut Microbiome Correlates of Response and Toxicity Following anti-CD19 CAR T Cell Therapy. Nat. Med. 2022, 28, 713–723. [CrossRef]
[162] Wind, T.T.; Gacesa, R.; Vich Vila, A.; de Haan, J.J.; Jalving, M.; Weersma, R.K.; Hospers, G.A.P. Gut Microbial Species and Metabolic Pathways Associated with Response to Treatment with Immune Checkpoint Inhibitors in Metastatic Melanoma. Melanoma Res. 2020, 30, 235. [CrossRef] [PubMed]
[163] Yin, H.; Yang, L.; Peng, G.; Yang, K.; Mi, Y.; Hu, X.; Hao, X.; Jiao, Y.; Wang, X.; Wang, Y. The Commensal Consortium of the Gut Microbiome Is Associated with Favorable Responses to Anti-Programmed Death Protein 1 (PD-1) Therapy in Thoracic Neoplasms. Cancer Biol. Med. 2021, 18. [CrossRef] [PubMed]
[164] Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, J.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti–PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [CrossRef]
[165] Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut Microbiome Affects the Response to anti-PD-1 Immunotherapy in Patients with Hepatocellular Carcinoma. J. Immunother. Cancer 2019, 7. [CrossRef]
[166] Botticelli, A.; Vernocchi, P.; Marini, F.; Quagliariello, A.; Cerbelli, B.; Reddel, S.; Chierico, F.D.; Pietro, F.D.; Giusti, R.; Tomassini, A.; et al. Gut Metabolomics Profiling of Non-Small Cell Lung Cancer (NSCLC) Patients Under Immunotherapy Treatment. J. Transl. Med. 2020, 18. [CrossRef]
[167] Binda, S.; Hill, C.; Johansen, E.; Obis, D.; Pot, B.; Sanders, M.E.; Tremblay, A.; Ouwehand, A.C. Criteria to Qualify Microorganisms as “Probiotic” in Foods and Dietary Supplements. Front. Microbiol. 2020, 11. [CrossRef]
[168] Ma, J.; Lyu, Y.; Liu, X.; Jia, X.; Cui, F.; Wu, X.; Deng, S.; Yue, C. Engineered Probiotics. Microb. Cell Factories 2022, 21, 72. [CrossRef]
[169] Suez, J.; Zmora, N.; Segal, E.; Elinav, E. The Pros, Cons, and Many Unknowns of Probiotics. Nat. Med. 2019, 25, 716–729. [CrossRef] [PubMed]
[170] Zhang, Z.; Gao, Q.; Ren, X.; Luo, M.; Liu, Y.; Liu, P.; Liu, Y.; Ye, Y.; Chen, X.; Liu, H.; et al. Characterization of Intratumor Microbiome in Cancer Immunotherapy. The Innovation 2023, 4, 100482. [CrossRef]
[171] Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti–PD-L1 Efficacy. Science 2015, 350, 1084–1089. [CrossRef]
[172] Lee, H.A.; Kim, H.; Lee, K.-W.; Park, K.-Y. Dead Nano-Sized Lactobacillus Plantarum Inhibits Azoxymethane/Dextran Sulfate Sodium-Induced Colon Cancer in Balb/c Mice. J. Med. Food 2015, 18, 1400–1405. [CrossRef]
[173] Talero, E.; Bolivar, S.; Ávila-Román, J.; Alcaide, A.; Fiorucci, S.; Motilva, V. Inhibition of Chronic Ulcerative Colitis-Associated Adenocarcinoma Development in Mice by VSL#3. Inflamm. Bowel Dis. 2015, 21, 1027–1037. [CrossRef]
[174] Liu, J.; Zhang, Y. Intratumor Microbiome in Cancer Progression: Current Developments, Challenges and Future Trends. Biomark. Res. 2022, 10. [CrossRef]
[175] Yang, W.; Chen, C.-H.; Jia, M.; Xing, X.; Gao, L.; Tsai, H.-T.; Zhang, Z.; Liu, Z.; Zeng, B.; Yeung, S.-C.J.; et al. Tumor-Associated Microbiota in Esophageal Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
[176] Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human Oral Microbiome and Prospective Risk for Pancreatic Cancer: A Population-Based Nested Case-Control Study. Gut 2018, 67, 120–127. [CrossRef]
[177] Noci, V.L.; Guglielmetti, S.; Arioli, S.; Camisaschi, C.; Bianchi, F.; Sommariva, M.; Storti, C.; Triulzi, T.; Castelli, C.; Balsari, A.; et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Rep. 2018, 24, 3528–3538. [CrossRef]
[178] Shi, Y.; Zheng, W.; Yang, K.; Harris, K.G.; Ni, K.; Xue, L.; Lin, W.; Chang, E.B.; Weichselbaum, R.R.; Fu, Y.-X. Intratumoral Accumulation of Gut Microbiota Facilitates CD47-Based Immunotherapy via STING Signaling. J. Exp. Med. 2020, 217, e20192282. [CrossRef]
[179] Zhang, M.; Eshraghian, E.A.; Jammal, O.A.; Zhang, Z.; Zhu, X. CRISPR Technology: The Engine That Drives Cancer Therapy. Biomed. Pharmacother. 2021, 133. [CrossRef]
[180] Merenstein, D.; Pot, B.; Leyer, G.; Ouwehand, A.C.; Preidis, G.A.; Elkins, C.A.; Hill, C.; Lewis, Z.T.; Shane, A.L.; Zmora, N.; et al. Emerging Issues in Probiotic Safety: 2023 Perspectives. Gut Microbes 2023, 15. [CrossRef]
[181] Secher, T.; Kassem, S.; Benamar, M.; Bernard, I.; Boury, M.; Barreau, F.; Oswald, E.; Saoudi, A. Oral Administration of the Probiotic Strain Escherichia coli Nissle 1917 Reduces Susceptibility to Neuroinflammation and Repairs Experimental Autoimmune Encephalomyelitis-Induced Intestinal Barrier Dysfunction. Front. Immunol. 2017, 8. [CrossRef]
[182] Zhang, Y.; Zhang, Y.; Xia, L.; Zhang, X.; Ding, X.; Yan, F.; Wu, F. Escherichia coli Nissle 1917 Targets and Restrains Mouse B16 Melanoma and 4T1 Breast Tumors Through Expression of Azurin Protein. Appl. Environ. Microbiol. 2012, 78, 7603–7610. [CrossRef]
[183] Mahdizade Ari, M.; Dadgar, L.; Elahi, Z.; Ghanavati, R.; Taheri, B. Genetically Engineered Microorganisms and Their Impact on Human Health. Int. J. Clin. Pract. 2024, 2024, 6638269. [CrossRef]
[184] Canale, F.P.; Basso, C.; Antonini, G.; Perotti, M.; Li, N.; Sokolovska, A.; Neumann, J.; James, M.J.; Geiger, S.; Jin, W.; et al. Metabolic Modulation of Tumours with Engineered Bacteria for Immunotherapy. Nature 2021, 598, 662–666. [CrossRef]
[185] Zhang, Y.; Tan, W.; Sultonova, R.D.; Nguyen, D.-H.; Zheng, J.H.; You, S.-H.; Rhee, J.H.; Kim, S.; Khim, K.; Hong, Y.; et al. Synergistic Cancer Immunotherapy Utilizing Programmed Salmonella typhimurium Secreting Heterologous Flagellin B Conjugated to Interleukin-15 Proteins. Biomaterials 2023, 298. [CrossRef]
[186] Mizel, S.B.; Bates, J.T. Flagellin as an Adjuvant: Cellular Mechanisms and Potential. J. Immunol. 2010, 185, 5677–5682. [CrossRef]
[187] Cui, B.; Liu, X.; Fang, Y.; Zhou, P.; Zhang, Y.; Wang, Y. Flagellin as a Vaccine Adjuvant. Expert Rev. Vaccines 2018, 17, 335–349. [CrossRef]
[188] Boyiadzis, M.; Memon, S.; Carson, J.; Allen, K.; Szczepanski, M.J.; Vance, B.A.; Dean, R.; Bishop, M.R.; Gress, R.E.; Hakim, F.T. Up-regulation of NK Cell Activating Receptors Following Allogeneic Hematopoietic Stem Cell Transplantation Under a Lymphodepleting Reduced Intensity Regimen Is Associated with Elevated IL-15 Levels. Biol. Blood Marrow Transplant. 2008, 14, 290–300. [CrossRef]
[189] ‘Mac’ Cheever, M.A. Twelve Immunotherapy Drugs That Could Cure Cancers. Immunol. Rev. 2008, 222, 357–368. [CrossRef]
[190] Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [CrossRef]
[191] Wei, C.; Xun, A.Y.; Wei, X.X.; Yao, J.; Wang, J.Y.; Shi, R.Y.; Yang, G.H.; Li, Y.X.; Xu, Z.L.; Lai, M.G.; et al. Bifidobacteria Expressing Tumstatin Protein for Antitumor Therapy in Tumor-Bearing Mice. Technol. Cancer Res. Treat. 2016, 15, 498–508. [CrossRef]
[192] Leventhal, D.S.; Sokolovska, A.; Li, N.; Plescia, C.; Kolodziej, S.A.; Gallant, C.W.; Christmas, R.; Gao, J.-R.; James, M.J.; Abin-Fuentes, A.; et al. Immunotherapy with Engineered Bacteria by Targeting the STING Pathway for Anti-Tumor Immunity. Nat. Commun. 2020, 11, 2739. [CrossRef]
[193] Gurbatri, C.R.; Lia, I.; Vincent, R.; Coker, C.; Castro, S.; Treuting, P.M.; Hinchliffe, T.E.; Arpaia, N.; Danino, T. Engineered Probiotics for Local Tumor Delivery of Checkpoint Blockade Nanobodies. Sci. Transl. Med. 2020, 12, eaax0876. [CrossRef]
[194] He, L.; Yang, H.; Tang, J.; Liu, Z.; Chen, Y.; Lu, B.; He, H.; Tang, S.; Sun, Y.; Liu, F.; et al. Intestinal Probiotics E. coli Nissle 1917 as a Targeted Vehicle for Delivery of p53 and Tum-5 to Solid Tumors for Cancer Therapy. J. Biol. Eng. 2019, 13. [CrossRef]
[195] Pan, H.; Li, L.; Pang, G.; Han, C.; Liu, B.; Zhang, Y.; Shen, Y.; Sun, T.; Liu, J.; Chang, J.; et al. Engineered NIR Light-Responsive Bacteria as Anti-Tumor Agent for Targeted and Precise Cancer Therapy. Chem. Eng. J. 2021, 426, 130842. [CrossRef]
[196] Riedel, C.U.; Casey, P.G.; Mulcahy, H.; O’Gara, F.; Gahan, C.G.M.; Hill, C. Construction of p16Slux, a Novel Vector for Improved Bioluminescent Labeling of Gram-Negative Bacteria. Appl. Environ. Microbiol. 2007, 73, 7092–7095. [CrossRef]
[197] Danino, T.; Prindle, A.; Kwong, G.A.; Skalak, M.; Li, H.; Allen, K.; Hasty, J.; Bhatia, S.N. Programmable Probiotics for Detection of Cancer in Urine. Sci. Transl. Med. 2015, 7, 289ra84. [CrossRef]
[198] Ali, A.; Ara, A.; Kashyap, M.K. Gut Microbiota: Role and Association with Tumorigenesis in Different Malignancies. Mol. Biol. Rep. 2022, 49, 8087–8107. [CrossRef]
[199] Han, K.; Nam, J.; Xu, J.; Sun, X.; Huang, X.; Animasahun, O.; Achreja, A.; Jeon, J.H.; Pursley, B.; Kamada, N.; et al. Generation of Systemic Antitumour Immunity via the in situ Modulation of the Gut Microbiome by an Orally Administered Inulin Gel. Nat. Biomed. Eng. 2021, 5, 1377–1388. [CrossRef]
[200] Hiraizumi, M.; Perry, N.T.; Durrant, M.G.; Soma, T.; Nagahata, N.; Okazaki, S.; Athukoralage, J.S.; Isayama, Y.; Pai, J.J.; Pawluk, A.; et al. Structural Mechanism of Bridge RNA-Guided Recombination. Nature 2024, 630, 994–1002. [CrossRef]
[201] Durrant, M.G.; Perry, N.T.; Pai, J.J.; Jangid, A.R.; Athukoralage, J.S.; Hiraizumi, M.; McSpedon, J.P.; Pawluk, A.; Nishimasu, H.; Konermann, S.; et al. Bridge RNAs Direct Programmable Recombination of Target and Donor DNA. Nature 2024, 630, 984–993. [CrossRef]