
 ACCESS
Perspective
ACCESS
Perspective
Volume 2, Article ID: 2025.0012

Gerson D. KEPPEKE
gerson.keppeke@ucn.cl

1 Departamento de Ciencias Biomédicas, Facultad de Medicina, Universidad Católica del Norte, Coquimbo 1781421, Chile
2 Disciplina de Reumatologia, Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, Brasil
Received: 05 Oct 2024 Accepted: 08 Jan 2025 Available Online: 10 Jan 2025 Published: 28 Feb 2025
This manuscript examines advancements in antigen-specific immunosuppression, as well as the potential and challenges of applying gene-editing technologies to autoimmune diseases driven by autoantibodies (AAbs). Current approved treatments fail to reach long-lived plasma cells (LLPCs), which may continue secreting pathogenic AAbs after immunobiological courses in some autoimmune illnesses. New approaches, some tested in vitro and some already undergoing clinical trials, such as the chimeric autoantibody receptor (CAAR)-T cells, BiTEs, affinity matrices, and CRISPR-AAb-untargeting, show promise in selectively eliminating autoreactive B lymphocytes. Challenges remain, particularly in adapting CRISPR for in vivo use. The future of precision medicine for autoimmunity will rely in AI-guided personalized approaches, enabling the design of highly specific therapies to target disease-causing autoreactive cells while preserving immune function, marking a transformative step in autoimmune treatment.
Many autoimmune diseases are characterized by the presence of autoantibodies (AAbs), which play key roles in the pathogenesis of these conditions. These autoantibodies can serve as both diagnostic biomarkers and direct effectors of tissue damage, as seen in diseases like Myasthenia Gravis, type I diabetes, Pemphigus Vulgaris, and autoimmune encephalopathies [1]. In many of these diseases, pathogenic AAbs bind to critical antigens in the body, interfering with normal biological functions and activating antibody-dependent immune mechanisms, leading to tissue destruction and perpetuation of the inflammatory cycle. The antigenic specificity of any antibody is determined by V(D)J gene rearrangements that occur during B cell development in the bone marrow. However, strong activation of these cells by cooperation with T-CD4+ cells and the subsequent formation of germinal centers in lymph nodes, results in the generation of long-lived plasma cell clones (LLPCs), which continue secreting high-affinity immunoglobulins even in the absence of antigenic stimuli [2]. Currently, standard therapies for autoimmune diseases include immunosuppressants, general B-cell depleting agents, complement inhibitors and FcRn antagonists, among others, such as the well-known anti-CD20 monoclonal antibodies (Figure 1). However, these therapies fail to target LLPCs, which do not depend on antigens to continue producing AAbs. In fact, there are very few therapies that can target LLPCs, such as the anti-CD38 antibody daratumumab, which has shown efficacy in targeting plasma cells, including LLPCs, and has been associated with reduced disease activity in systemic lupus erythematosus (SLE) [3]. Additionally, other interventions like plasmapheresis and FcRn antagonists are only temporary, as AAbs levels quickly return due to the persistence of LLPCs, and autologous bone marrow transplant is usually the last resort in autoimmune diseases due to the risks of opportunistic infections associated with the procedure. Other recent experimental approaches, such as using cytolytic cells (CD8+ T cells) transduced with chimeric autoantibody receptors (CAAR-T) [4], have demonstrated some success in eliminating antigen-specific B cells but still do not fully address the problem of LLPCs, which are the primary source of approximately half of the circulating antibodies in an adult (Figure 1). Eliminating antigen-specific LLPCs is the ultimate therapeutic goal for managing pathogenic AAb-driven autoimmune diseases. However, LLPCs pose significant challenges for targeted therapies due to their unique survival mechanisms and protective microenvironments. These cells can persist for decades, producing antibodies that contribute to autoimmunity, graft rejection, and drug neutralization [5]. Residing in specialized niches within the bone marrow, LLPCs are supported by mesenchymal and hematopoietic components that provide essential survival factors, such as interleukin-6 (IL-6), tumor necrosis factor (TNF), and anti-apoptotic signals like Mcl-1 [6]. These niches, combined with intrinsic programs like enhanced autophagy, metabolic fitness, and stress response mechanisms, allow LLPCs to resist conventional therapies, including immunosuppression, B cell depletion, and irradiation [7]. Unlike short-lived plasma cells, LLPCs remain quiescent and do not express common activation markers like CD20, rendering them invisible to most therapeutic approaches. Moreover, LLPCs secrete all their produced antibodies without retaining them as membrane-bound B-cell receptors (BCRs), which limits the effectiveness of therapies like CAAR-T cells [8]. A recent breakthrough deserving attention is the use of bispecific T-cell engagers (BiTEs) in the treatment of autoimmune diseases (Figure 1), particularly conditions mediated by the B-cells and their progeny, the plasma cells, such as SLE and rheumatoid arthritis (RA). BiTEs are essentially molecules comprising two Ab variable regions targeting two receptors, such as CD3 and CD19, designed to force the interaction between the two cell populations involved [9]. Bucci et al. [10] demonstrated the efficacy of blinatumomab, a CD19xCD3 BiTE, in patients with multi-drug-resistant RA, achieving significant B cell depletion, reduced disease activity, and improved synovitis [10]. Originally developed for oncology, BiTEs like blinatumomab have revolutionized cancer immunotherapy, particularly in hematologic malignancies such as acute lymphoblastic leukemia [11]. The modular design of BiTEs allows the targeting of diverse antigens, enabling the development of personalized therapies for both oncology and immunology. The success of teclistamab, a BCMAxCD3 BiTE that directs T cells against plasma cells, in treating multiple myeloma further underscores the adaptability of this platform for precision immunotherapy, including some autoimmune diseases [12]. For example, a recent in vivo trial applied teclistamab in a patient with SLE, resulting in disease remission [13]. These findings open promising avenues for repurposing BiTEs to manage autoimmune diseases, with a specific focus on targeting pathogenic B cells [14]. Given the resistance of LLPCs to conventional therapies, there is an urgent need for innovative, preferably antigen-specific interventions, which can selectively eliminate or modulate pathogenic plasma cell clones secreting AAbs. In this context, a recent in vitro study proposed the use of CRISPR/Cas9 gene-editing technology as a tool to target the V(D)J rearrangements responsible for the production of pathogenic autoantibodies [15]. By disrupting given rearrangements in the genes encoding immunoglobulins, it is possible to eliminate the ability of LLPCs to produce AAbs, paving the way for a highly specific and precise therapy. In the in vitro study by our group [15], we demonstrated that the CRISPR/Cas9 system is indeed effective in knocking out autoantibodies by targeting the V(D)J rearrangements. Even when complete knockout of the gene was not achieved, point mutations inserted after the nuclease Cas cut at the sites encoding the CDRs significantly affected and nearly eliminated the ability of the AAbs to bind to their target. This CRISPR-based untargeting approach shows promise for reducing the quantity and/or specificity of AAbs and could be adapted for potential in vivo applications [15]. This innovative approach, which aims to directly deactivate the genes that produce the disease-causing AAbs, could represent a paradigm shift in the treatment of AAb-mediated autoimmune diseases. The objective in this manuscript was to discuss the current context related to antigen-specific immunosuppression and the main challenges concerning the adaptation of the CRISPR-AAb-untargeting system for in vivo application.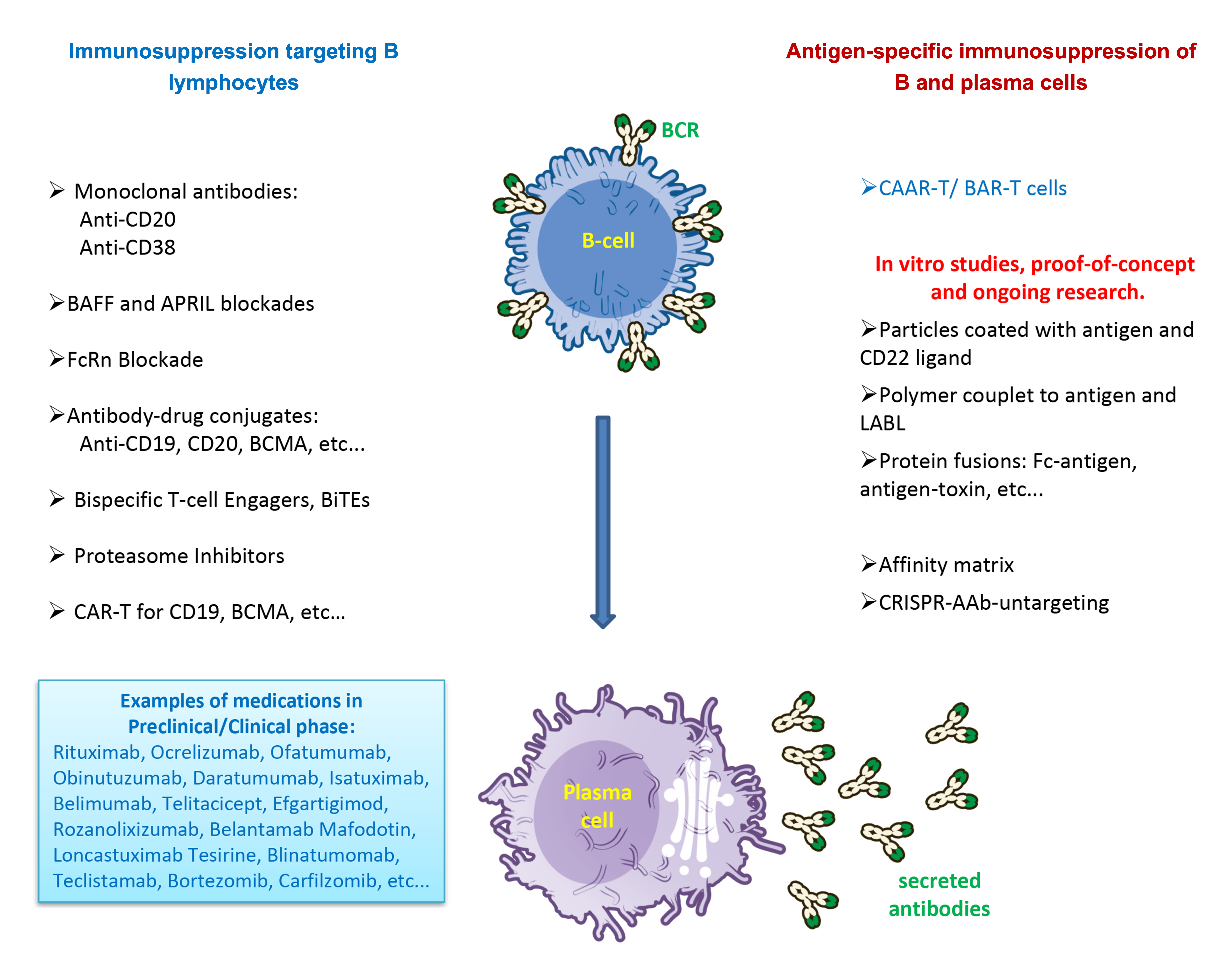
Despite the current challenges in adapting the CRISPR system for in vivo therapies—such as delivering the Cas9 enzyme and gRNA machinery to the target tissue cells requiring editing—CRISPR-based therapies for sickle cell anemia (SCD) and beta-thalassemia have recently received FDA approval [16]. In addition, concerns about off-target effects and immune responses persist. Frati et al. [17] observed more pronounced off-target activity and chromosomal rearrangements in SCD samples compared to healthy donors, emphasizing the need for disease-specific safety assessments. Additionally, upregulation of DNA damage and inflammatory response genes in edited cells was noted [17]. Lopes & Prasad [18] stress in a recent review the importance of comprehensive off-target detection methods and improved CRISPR systems to enhance therapeutic safety [18]. Despite these challenges, gene-edited SCD hematopoietic stem and progenitor cells (HSPCs) have shown successful results reducing sickling of red blood cells in SCD patients, suggesting potential clinical benefits if safety concerns can be adequately addressed [16]. The approved CRISPR-based therapies use an ex vivo strategy, where HSPCs are extracted from the patients, and after editing to knockout the beta-hemoglobin gene, the cells are selected, expanded, and reinjected into the same patient. In the case of gene editing to eliminate pathogenic V(D)J rearrangements [15], i.e., AAbs, this approach could be adapted in a similar manner, as LLPCs reside in the bone marrow and could be extracted, edited, and reinjected into patients with the pool of LLPCs no longer secreting the pathogenic AAbs. The major problem or current challenge is that autoantibodies are polyclonal, meaning that multiple V(D)J rearrangements generate antibodies with distinct variable regions, but that bind at the same target, although with varying affinities, i.e., bind at different (or similar) epitopes that are all parts of the same antigen. In this context, defining which pathogenic V(D)J rearrangements should be targeted by the gRNAs for knockout by Cas9 becomes a daunting task, considering that an adult has approximately 1 million B-cell clones, i.e., plasma cells clones secreting antibodies [15]. To overcome this obstacle, a promising approach is the use of Artificial Intelligence (AI) to predict the epitopes to which the AAbs generated by specific V(D)J rearrangements will bind. In the past three years, hybrid models based on neural networks and machine learning, integrating supervised and unsupervised learning, have emerged. These models are capable of analyzing deep sequencing data from variable antibody regions, predicting the conformation of paratopes and their complementarity-determining regions (CDRs) with high confidence, and accurately identifying potential target epitopes [19,20,21]. Using next-generation sequencing (NGS) to obtain the variable regions of antibodies from a given patient, these software tools could analyze and identify common patterns that might represent the pool of pathogenic autoantibodies for that specific patient. This approach enables the selection of common sites that could be targeted by gRNAs, acting specifically on the most pathogenic rearrangements. This would optimize the editing process and increase precision in eliminating the genes responsible for producing AAbs in LLPCs, especially in more severe autoimmune conditions [19]. The use of autoregressive neural networks has already proven efficient in predicting the probability of high-affinity antibody-antigen binding. These models are trained on large sequencing data libraries, allowing millions of paratopes to be generated and tested in silico. This way, highly specific and effective sequences can be prioritized for experimental testing, selecting rearrangements with greater pathogenic potential. These methods are particularly useful in co-optimizing multiple antibody properties, such as affinity and specificity, while minimizing the impact of non-specific binding [20,21,22]. However, caution is warranted when relying solely on AI-based antibody predictions. A study on SARS-CoV-2 antibodies found that AI-predicted epitopes were inaccurate when compared to experimental validation [23]. Despite limitations, AI-driven approaches continue to evolve, offering potential time and cost reductions in antibody discovery pipelines [24]. While AI shows promise in enhancing antibody development processes, current models may lack the clinical robustness required for immediate therapeutic applications, highlighting the need for extensive wet-lab experimental validation alongside computational predictions. Additionally, algorithms like AlphaFold, OpenFold, OmegaFold, EsmFold, etc... which predict protein structure with high accuracy, can be used to improve the prediction of interactions between epitopes and AAbs. However, although AlphaFold is effective in predicting the structure of antigens, it is still limited in predicting conformation of specific antibody regions, such as the CDR-containing variable region. The improvement of docking techniques between antibodies and antigens, along with advances in neural networks focused on analyzing antibodies structures and their maturation-associated mutations, such as IgFold, DeepAb, RosettaAntibody and AbPredict, to mention just a few, has the potential to further refine the choice of gRNAs targets for V(D)J editing, ensuring a highly precise and effective therapeutic approach [19,20,25]. Despite the potential advances by associating AI-based antibody predictions and genome editing, safety concerns should be considered, particularly immunogenicity, which remains a major obstacle for in vivo CRISPR applications, necessitating careful design and mitigation strategies [26]. Ethical considerations surrounding CRISPR use are complex, encompassing issues such as permissible applications, equitable access, and the need for regulatory frameworks, especially for germline editing [27]. While the first CRISPR-based therapeutics have received regulatory approval [16], long-term effects remain unknown, and pivotal clinical trials for emerging applications are expected to be concluded within the next 5 years. As CRISPR technology advances, ongoing research, long-term patient follow-up, and evolving moral decision-making will be crucial to address safety concerns and ethical implications, ultimately determining the feasibility and scope of clinical implementation [27,28].
Recent developments in antigen-specific therapies targeting autoreactive B cells have shown great promise in treating autoimmune diseases, particularly because the outcome is reduction of circulating pathogenic This approach targets autoantibodies (AAbs) without requiring generalized immune suppression [29] (Figure 1). A notable example is the use of CAAR-T cells, a technology where cytolytic T cells are modified to express receptors with extracellular autoantigens to which autoreactive BCRs bind. For example, CAAR-T cells have been successful in diseases like pemphigus vulgaris, targeting B cells with BCRs, and in extension the plasma cells, that produce antibodies against desmoglein-3, a crucial protein for skin integrity [4,30]. This antigen-specific approach has proven effective in selectively depleting pathogenic B cells while preserving healthy, non-reactive B cells, representing a significant advance over conventional B cell depletion therapies like anti-CD20 monoclonal antibodies. In addition to CAAR-T cells, other strategies include affinity matrices, Fc-antigen fusion proteins, soluble multivalent antigen arrays, and particle-based therapeutics (Figure 1). A comprehensive review of the topic can be found elsewhere [29]. Two examples are mentioned here: The affinity matrix works by linking the recombinant antigen of interest to an antibody targeting plasma cell surface markers such as CD138 or CD44. This complex binds to plasma cells producing AAbs against the antigen of interest. The Fc region of the AAbs in turn recruits the complement system, inducing complement-dependent cytotoxicity to eliminate these cells, like a complement-dependent suicide [31]. Soluble multivalent antigen arrays work by coupling autoantigens to a polymeric backbone along with cell adhesion inhibitory peptides (LABL). These arrays directly bind to BCRs specific for the antigen, rendering B cells anergic or non-responsive. This method has shown efficacy in experimental models, such as autoimmune encephalomyelitis, preventing the activation of pathogenic B cells and inducing immune tolerance [32,33]. Among the various concepts of antigen-specific immunosuppression, as discussed elsewhere [29], the only ones capable of targeting LLPCs specifically are the affinity matrix and the CRISPR-AAb- untargeting system [15] (Figure 1). However, eliminating B cells that have not yet secreted AAbs can also indirectly affect plasma cells by reducing the number of new autoreactive antibody-secreting cells maturing from activated B lymphocytes. One could test the strategy to apply antigen-specific B-cell immunosuppression, such as the CAAR-T, together with traditional chimeric antigen receptor (CAR) T cells design to eliminate overall plasma cells, such as the anti-BCMA-CAR-T, as discussed elsewhere [34]. Alternatively, CAAR-T could be combined with the BCMAxCD3 BiTE teclistamab for enhanced effectiveness. Although these are not antigen-specific and affect all plasma cells, they could reduce the population of cells secreting AAbs while maintaining a system where new, non-autoreactive B cells, which are not eliminated by CAAR-T, could repopulate the patient’s immune system and plasma cells without the cells secreting pathogenic AAbs. It is worth noting that CAAR-T therapy has been successful in some diseases where short-lived plasma cells are the primary source of AAbs [30,35].
I envision a future where, through the analysis of variable regions of a patient’s antibodies via NGS, a personalized “antibodyomics” approach utilizing AI could identify the CDRs responsible for generating AAbs with the highest pathogenic potential. The same AI could be used to determine potential sequences for targeting through gene-editing techniques, such as CRISPR. Using a set of tools, including CAAR-T, affinity matrix, CRISPR-AAb-untargeting, and others, a patient with an AAb-driven autoimmune disease could undergo antigen-specific immunosuppression. This approach would eliminate both B cells with autoreactive BCRs and short- and long-lived plasma cells that secrete the respective pathogenic autoantibodies, representing an advanced concept of super precision medicine. Antigen-specific immune activation was successfully achieved about a century ago with vaccines, revolutionizing human medicine and health, but antigen-specific immunosuppression of B/T cells and their progeny is still in its early days, with great future prospects. Essentially this is the ultimate therapeutic goal for managing pathogenic AAb-driven autoimmune diseases.
AAb
Autoantibody
AI
Artificial Intelligence
BiTE
Bispecific T-cell Engager
BCR
B-cell Receptor
CAR-T
Chimeric Antigen Receptor T-cell
CAAR-T
Chimeric Autoantibody Receptor T-cell
CD
Cluster of Differentiation
CDR
Complementarity-Determining Region
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
FDA
Food and Drug Administration
FcRn
Neonatal Fc Receptor
gRNA
Guide RNA for CRISPR/Cas
HSPCs
Hematopoietic Stem and Progenitor Cells
IL
Interleukin
LLPC
Long-Lived Plasma Cell
ML
Machine Learning
NGS
Next-Generation Sequencing
RA
Rheumatoid Arthritis
SCD
Sickle Cell Disease
SLE
Systemic Lupus Erythematosus
TNF
Tumor Necrosis Factor
The author confirms to be solely responsible for the conception, design, analysis, interpretation, drafting, and final approval of the article.
Not applicable.
Not applicable.
The author declares no conflicts of interest regarding this manuscript.
G.D.K. is/was supported by Sao Paulo State Research Foundation, FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo). Grant numbers: “#2017/20745-1; #2023/07698-5; #2023/17946-6”.
The manuscript was originally written in Portuguese, the mother language of the author(s), and translated to English using online tools such as ChatGTP (by OpenAI) and Gemini (by Google/Alphabet). The final text underwent proof by an English language expert.
[1] R.J. Ludwig, K. Vanhoorelbeke, F. Leypoldt, Z. Kaya, K. Bieber, S.M. McLachlan, et al., "Mechanisms of Autoantibody-Induced Pathology" Front. Immunol., vol. 8, 2017. [Crossref] [PubMed]
[2] B.T. Gaudette, D. Allman, "Biochemical coordination of plasma cell genesis" Immunol. Rev., vol. 303, pp. 52-61, 2021. [Crossref] [PubMed]
[3] M. Yalcin Mutlu, J. Wacker, K. Tascilar, J. Taubmann, B. Manger, G. Kronke, et al., "Effective and safe treatment of anti-CD38 therapy in systemic lupus erythematosus-associated refractory cerebral vasculitis induces immune tolerance" Rheumatology, vol. 62, pp. e21-e23, 2023. [Crossref]
[4] J. Lee, D.K. Lundgren, X. Mao, S. Manfredo-Vieira, S. Nunez-Cruz, E.F. Williams, et al., "Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris" J. Clin. Investig., vol. 130, pp. 6317-6324, 2020. [Crossref] [PubMed]
[5] C. Markmann, V.G. Bhoj, "On the road to eliminating long-lived plasma cells-“are we there yet?”" Immunol. Rev., vol. 303, pp. 154-167, 2021. [Crossref]
[6] O. Winter, C. Dame, F. Jundt, F. Hiepe, "Pathogenic long-lived plasma cells and their survival niches in autoimmunity, malignancy, and allergy" J. Immunol., vol. 189, pp. 5105-5111, 2012. [Crossref] [PubMed]
[7] A. Utley, B. Lipchick, K.P. Lee, M.A. Nikiforov, "Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells" Cancers, vol. 12, 2020. [Crossref] [PubMed]
[8] S.M. Lightman, A. Utley, K.P. Lee, "Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle" Front. Immunol., vol. 10, 2019. [Crossref] [PubMed]
[9] F. Humby, B. Kirkham, L. Taams, "BiTE therapy for rheumatoid arthritis" Nat. Med., vol. 30, pp. 1533-1534, 2024. [Crossref] [PubMed]
[10] L. Bucci, M. Hagen, T. Rothe, M.G. Raimondo, F. Fagni, C. Tur, et al., "Bispecific T cell engager therapy for refractory rheumatoid arthritis" Nat. Med., vol. 30, pp. 1593-1601, 2024. [Crossref] [PubMed]
[11] D. Deak, C. Pop, A.A. Zimta, A. Jurj, A. Ghiaur, S. Pasca, et al., "Let’s Talk About BiTEs and Other Drugs in the Real-Life Setting for B-Cell Acute Lymphoblastic Leukemia" Front. Immunol., vol. 10, 2019. [Crossref] [PubMed]
[12] H. Einsele, H. Borghaei, R.Z. Orlowski, M. Subklewe, G.J. Roboz, G. Zugmaier, et al., "The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types" Cancer, vol. 126, pp. 3192-3201, 2020. [Crossref] [PubMed]
[13] T. Alexander, J. Kronke, Q. Cheng, U. Keller, G. Kronke, "Teclistamab-Induced Remission in Refractory Systemic Lupus Erythematosus" N. Engl. J. Med., vol. 391, pp. 864-866, 2024. [Crossref]
[14] M. Hagen, L. Bucci, S. Boltz, D.M. Nothling, T. Rothe, K. Anoshkin, et al., "BCMA-Targeted T-Cell-Engager Therapy for Autoimmune Disease" N. Engl. J. Med., vol. 391, pp. 867-869, 2024. [Crossref]
[15] G.D. Keppeke, L. Diogenes, K. Gomes, L.E.C. Andrade, "“Untargeting” autoantibodies using genome editing, a proof-of-concept study" Clin. Immunol., vol. 251, p. 109343, 2023. [Crossref]
[16] N. Healey, "Next-generation CRISPR-based gene-editing therapies tested in clinical trials" Nat. Med., vol. 30, pp. 2380-2381, 2024. [Crossref] [PubMed]
[17] G. Frati, M. Brusson, G. Sartre, B. Mlayah, T. Felix, A. Chalumeau, et al., "Safety and efficacy studies of CRISPR-Cas9 treatment of sickle cell disease highlights disease-specific responses" Mol. Ther. J. Am. Soc. Gene Ther., vol. 32, pp. 4337-4352, 2024. [Crossref] [PubMed]
[18] R. Lopes, M.K. Prasad, "Beyond the promise: Evaluating and mitigating off-target effects in CRISPR gene editing for safer therapeutics" Front. Bioeng. Biotechnol., vol. 11, 2023. [Crossref] [PubMed]
[19] E.K. Makowski, H.T. Chen, P.M. Tessier, "Simplifying complex antibody engineering using machine learning" Cell Syst., vol. 14, pp. 667-675, 2023. [Crossref] [PubMed]
[20] M. Chungyoun, J.J. Gray, "AI Models for Protein Design are Driving Antibody Engineering" Curr. Opin. Biomed. Eng., vol. 28, 2023. [Crossref] [PubMed]
[21] J. Parkinson, R. Hard, W. Wang, "The RESP AI model accelerates the identification of tight-binding antibodies" Nat. Commun., vol. 14, p. 454, 2023. [Crossref] [PubMed]
[22] E.K. Makowski, P.C. Kinnunen, J. Huang, L. Wu, M.D. Smith, T. Wang, et al., "Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space" Nat. Commun., vol. 13, p. 3788, 2022. [Crossref]
[23] D.D. Acar, W. Witkowski, M. Wejda, R. Wei, T. Desmet, B. Schepens, et al., "Integrating artificial intelligence-based epitope prediction in a SARS-CoV-2 antibody discovery pipeline: Caution is warranted" EBioMedicine, vol. 100, 2024. [Crossref] [PubMed]
[24] J. Cheng, T. Liang, X.Q. Xie, Z. Feng, L. Meng, "A new era of antibody discovery: An in-depth review of AI-driven approaches" Drug Discov. Today, vol. 29, p. 103984, 2024. [Crossref] [PubMed]
[25] D.M. Mason, S. Friedensohn, C.R. Weber, C. Jordi, B. Wagner, S.M. Meng, et al., "Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning" Nat. Biomed. Eng., vol. 5, pp. 600-612, 2021. [Crossref] [PubMed]
[26] R. Ewaisha, K.S. Anderson, "Immunogenicity of CRISPR therapeutics-Critical considerations for clinical translation" Front. Bioeng. Biotechnol., vol. 11, 2023. [Crossref] [PubMed]
[27] C. Brokowski, M. Adli, "CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool" J. Mol. Biol., vol. 431, pp. 88-101, 2019. [Crossref] [PubMed]
[28] F. Memi, A. Ntokou, I. Papangeli, "CRISPR/Cas9 gene-editing: Research technologies, clinical applications and ethical considerations" Semin. Perinatol., vol. 42, pp. 487-500, 2018. [Crossref] [PubMed]
[29] Z.C. Stensland, J.C. Cambier, M.J. Smith, "Therapeutic Targeting of Autoreactive B Cells: Why, How, and When?" Biomedicines, vol. 9, 2021. [Crossref]
[30] A. Vafaeian, H. Mahmoudi, M. Daneshpazhooh, "What is novel in the clinical management of pemphigus vulgaris?" Expert Rev. Clin. Pharmacol., vol. 17, pp. 489-503, 2024. [Crossref] [PubMed]
[31] Q. Cheng, A. Pelz, A. Taddeo, L. Khodadadi, J. Klotsche, B.F. Hoyer, et al., "Selective depletion of plasma cells in vivo based on the specificity of their secreted antibodies" Eur. J. Immunol., vol. 50, pp. 284-291, 2020. [Crossref] [PubMed]
[32] B.L. Hartwell, F.J. Martinez-Becerra, J. Chen, H. Shinogle, M. Sarnowski, D.S. Moore, et al., "Antigen-Specific Binding of Multivalent Soluble Antigen Arrays Induces Receptor Clustering and Impedes B Cell Receptor Mediated Signaling" Biomacromolecules, vol. 17, pp. 710-722, 2016. [Crossref]
[33] B.L. Hartwell, C.J. Pickens, M. Leon, C. Berkland, "Multivalent Soluble Antigen Arrays Exhibit High Avidity Binding and Modulation of B Cell Receptor-Mediated Signaling to Drive Efficacy against Experimental Autoimmune Encephalomyelitis" Biomacromolecules, vol. 18, pp. 1893-1907, 2017. [Crossref] [PubMed]
[34] G. Schett, F. Muller, J. Taubmann, A. Mackensen, W. Wang, R.A. Furie, et al., "Advancements and challenges in CAR T cell therapy in autoimmune diseases" Nat. Rev. Rheumatol., vol. 20, pp. 531-544, 2024. [Crossref] [PubMed]
[35] S. Crunkhorn, "CAAR T cells to treat encephalitis" Nat. Rev. Drug Discov., vol. 23, p. 22, 2024. [Crossref]