
 ACCESS
Research Article
ACCESS
Research Article
Volume 1, Article ID: 2024.0002

Victor Furer
furer@kgasu.ru


Alexandr E. Vandyukov
sasha@iopc.ru
Ekaterina I. Nomerotskaya
nomkat@mail.ru
Mariyam M. Mukhtarova
mariyam-99@mail.ru
Vladimir V. Kovalev
kovalev@petrol.chem.msu.ru
Valery I. Kovalenko
koval@iopc.ru
1 Department of Physics, Electrical Engineering and Automation, Kazan State Architect and Civil Engineering University, 1 Zelenaya, 420043 Kazan, Russia
2 A.E. Arbuzov Institute of Organic and Physical Chemistry, RAS, 8 Arbuzov Str., 420088 Kazan, Russia
3 Department of Chemistry, Moscow State University, 1-3 Lenin’s Hills, 119991 Moscow, Russia
* Author to whom correspondence should be addressed
Received: 26 Sep 2024 Accepted: 06 Dec 2024 Available Online: 06 Dec 2024 Published: 26 Dec 2024
The heterocyclic structure of pyrones has a variety of biological activities and plays an important role in the creation of new drugs. Therefore, the study of the structure and spectra of pyrones is of considerable interest. In this work, the IR and Raman spectra of 3-(3,3-Dimethylbutanoyl)-4-hydroxy-6-neopentyl-2H-pyran-2-one (1) in its crystalline state was studied. The tautomerization of 1 was followed by a quantum-chemical method at the DFT/B3LYP/6-311G** level. The calculation for the 4-hydroxy enol tautomer (A) reproduces the experimental IR and Raman spectra of compound 1. The classification of the bands in the experimental vibrational spectra of 1 has been carried out. The intramolecular H-bond was characterized by IR spectroscopy. The free energies of the tautomers and their populations were calculated for two different solvents. It appears from our data that type A dominates. The content of tautomer B increases in the nonpolar solvent but does not exceed 13%. As evident from the calculations and experimental X-ray data, the pyran ring of the molecule is flat. HOMO and LUMO of molecule 1 are located on the pyran ring. During tautomeric transformations, there is a significant delocalization of charge and a change in the reactivity of the molecule. The reactivity of pyrone 1 was characterized using descriptors. The form B was found to have higher ionization energy, electron affinity, chemical potential, and electrophilic index than the A form. The dipole moment is higher for form A, and the softness of the two molecules is the same.
The study of heterocyclic pyrone derivatives is interesting and important because they are used in the pharmaceutical, cosmetic, and food industries [1,2,3,4,5]. Pyrones are biologically active substances and are used for the manufacture of analgesics, anti-cancer drugs, and to fight against HIV [1,2,3,4,5,6,7,8,9,10]. Pyrones are the initial reagents in the synthesis of many organic compounds [1,2,3,4,5,6,7,8,9,10]. Studies on NMR spectra have shown that among the five tautomeric forms of the pyrones, two enolic forms predominate [11,12]. The IR and NMR spectra of pyrones have been studied [13,14,15,16,17,18]. This particular work for the first time characterized the two low-energy tautomeric forms of the 3-(3,3-dimethylbutanoyl)-4-hydroxy-6-neopentyl-2H-pyran-2-one (1) using the methods of IR and Raman spectroscopy and quantum chemistry. The choice of compound 1 is linked to the tautomerism of the central structural fragment of pyrandione, which can modify certain features characteristic of the functional groups. The enamine derivative of synthesized pyrone 1 has inhibitory activity against the human carcinoma cell line HeLa and the herpes virus VPG [19]. We attempted to trace the change in the structure of the acid, the strength of the hydrogen bond and its vibrational spectra during tautomeric transformations. The comparison of the free energies of the tautomers allows for an estimation of their population. It was important to follow the evolution of the geometry, electronic structure, and spectra of pyrone 1 during tautomeric transformations. Active centers of the molecule for nucleophilic and electrophilic attacks have been determined. The calculation of the charges on the acid atoms made it possible to estimate the capacity of the atoms to form hydrogen bonds and attract ions and metal atoms. The electrophilicity index characterizes the biological activity of compound 1.
2.1. Experimental The neopentyl derivative of dehydroacetic acid 3-(3,3-dimethylbutanoyl)-4-hydroxy-6-neopentyl-2H-pyran-2-one (1) has been obtained by CF3SO3H/(CF3CO)2O activated acylation of carboxylic acids according to [19,20]. The white product has the crystalline powder form (melting point 77–78°C). Compound 1 can exist in two tautomeric enol forms, A and B (Figure 1). IR spectra were recorded by accumulating 64 scans in the region of 4000–400 cm−1 with a resolution of 4 cm−1. A Bruker Vector 22 spectrometer was used [21]. The samples were compressed into KBr pellets. Raman spectra of the pyrone were recorded in the 3500–50 cm−1 region via the FTIR spectrometer VERTEX 70 and the Bruker FT-Raman RAM II module [21]. The 1064 nm excitation line provided by an Nd:YAG laser with a power of 50 mW was used. 2.2. Computational Details The calculation of the vibrational spectra of compound 1 was performed using the B3LYP functional [22,23] and the basis set 6-311G**. The calculations were performed using the Gaussian09 program [24]. As a first approximation, the experimental coordinates of the atoms obtained through the X-ray diffraction method were used (Supplementary Information S1). Standard optimization methods were used to find minima on the potential surface. Full geometry optimization was performed without any restrictions. The Hessian analysis made it possible to determine the minima of potential energy. Optimized geometrical parameters of the tautomers were used to calculate the harmonic vibration frequencies. Theoretical structural and spectral data were obtained for both tautomers at 298 K, 1 atm. The potential energy distribution was calculated to attribute the vibrations [25]. Calculated frequencies were scaled using a multiplier of 0.96. The theoretical spectral curves were constructed, taking the Lorentz band shape and a half-width of 10 cm−1. The calculation of natural bonding orbitals (NBO) has been performed to characterize the electronic properties of molecules [26]. The chemical potential, hardness, softness, and electrophilicity index are related to the first vertical ionization energy and electron affinity by the following formulas: μ ≈ −(IE + EA)/2, η ≈ (IE − EA), S = 1/η, and ω = μ2/2η [27]. The Fukui functions for nucleophilic Using the difference in free energies of the tautomers, their populations at 298.15 K can be calculated 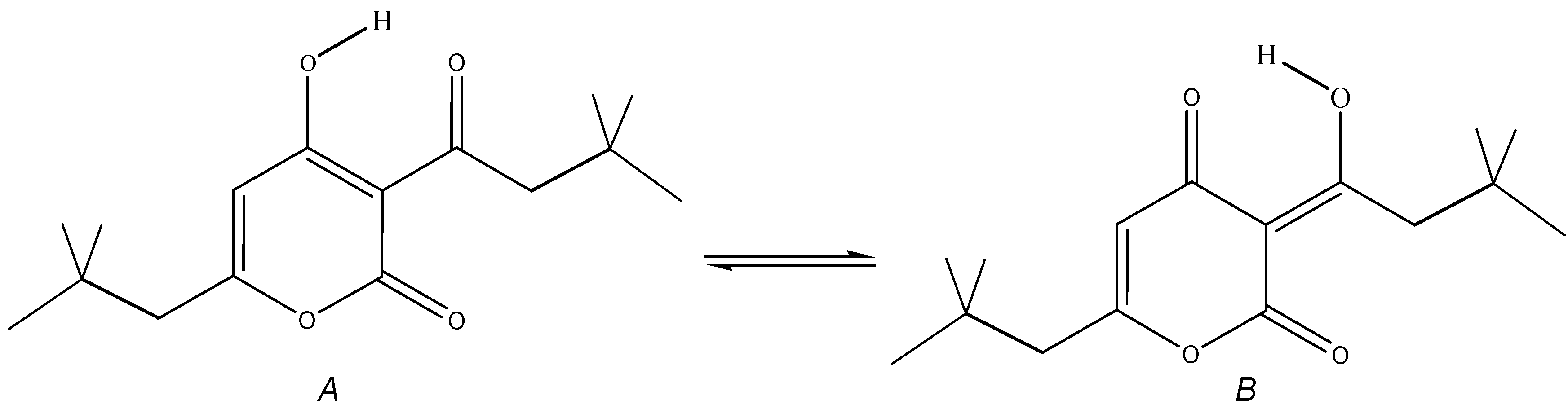
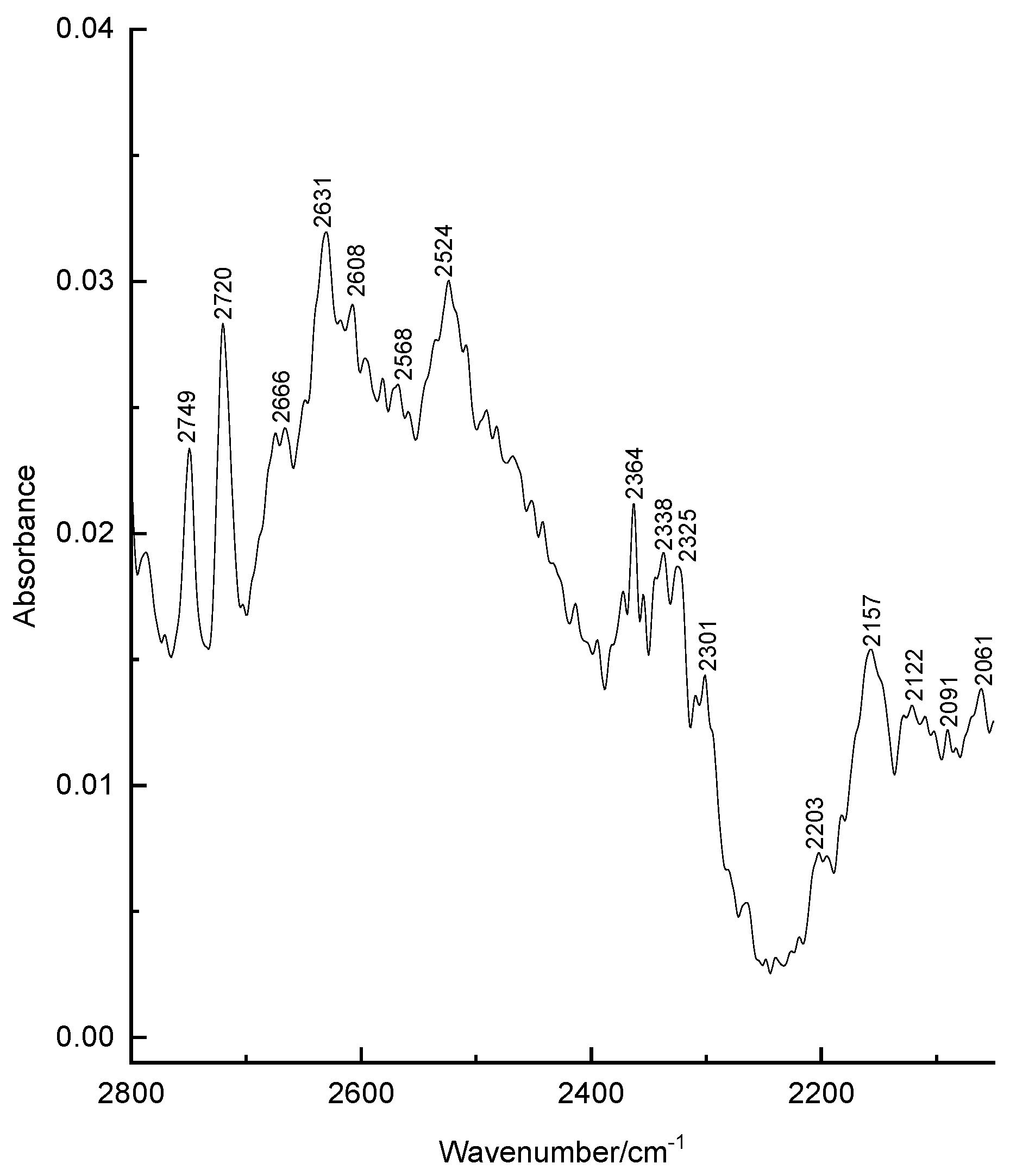
3.1. Structural Analysis As can be seen from the X-ray diffraction data in the crystalline state at room temperature, the A tautomer of compound 1 is realized (Supplementary Information S1). In the more stable tautomer A, an intramolecular hydrogen bond is realized. The measured distance between the O3 and O4 atoms is 2.437 Å. Supplementary Information S2 lists the measured bond lengths and angles of tautomer A. The results of geometry optimization of tautomers A and B are shown in Figure 2 and in Supplementary Information S2. Gibbs’s free energy and Boltzmann weights of tautomers are shown in Table 1. It appears from our data that type A dominates. The content of tautomer B increases in the less polar chloroform but does not exceed 13%. Vibrational spectra were calculated for tautomers A and B. As can be seen from the calculations and experimental X-ray data, the pyran ring of the molecule is flat. A satisfactory agreement is observed between the calculated geometrical parameters of tautomer A and the experimental X-ray data. Bond lengths change during tautomeric transformation. In tautomeric form A, the calculated bond lengths are (Å) 1.313 (O(3)–C(7)), 1.250 (O(4)–C(27)), 1.413 (C(6)–C(7)), and 1.467 (C(6)–C(27)), and for the B tautomer, the length of these bonds changes to 1.259, 1.302, 1.458, and 1.411, respectively. Such changes in bond lengths are consistent with the change in their properties during the tautomeric transformation. The H-bond lengths in the A and B tautomers are also different. The calculated O(3)∙∙∙O(4) distances for tautomers A and B are 2.473 and 2.418 Å, respectively. Gibbs relative free energy ΔG (kcal/mol), Boltzman weighting factor p (%) of low energy tautomers of 1 calculated in the B3LYP/6-311**G level. 3.2. Frontier Orbitals and Descriptors The HOMO and LUMO molecular orbitals for the A and B tautomers are located on the pyran ring (Figure 3). Conjugation leads to a flat structure for this ring. It is interesting to see how the charge distribution changes during tautomeric transformations. In the tautomeric transformation from form A to B, the negative charge on the O3 atom increases, and that on the O4 atom decreases (Supplementary Information S3). In the B form, the charges on the atoms C5, C6, and C7 increase, and on the C27, H44 atoms decrease. It follows from these data that the chemical properties change during the tautomeric transition. The pyrone 1 molecule contains several functional groups. Their reactions can be described using descriptors. Form B was found to have higher ionization energy, electron affinity, chemical potential, electrophilic index, and energy band gap than Form A (Table 2). The dipole moment is higher for form A, and the softness of both molecules is the same. Calculated ionization energy (I), electron affinity (A), energy band gap (|GAP|), chemical potential (μ), global softness (S), global electrophilicity index (ω), and dipole moment (M) for tautomeric forms A and B of compound 1. The calculation of the local electrophilic indices makes it possible to estimate the reactivity of the atoms for two tautomeric forms (Supplementary Information S3). The electrophilic index of oxygen atoms is higher for the A form. For the carbon atoms C5, C6, C7, and C27, the electrophilicity index is higher for form A. It should be emphasized that the C6 atom is highly reactive. Detailed analysis of chemical descriptors allows finding new ways to obtain drugs with desired properties. 3.3. NBO Analysis The molecular orbitals are similar for the two tautomeric forms, but there is an orbital σ(2)C6–C7 = 0.8137(sp1.00d0.00)C6 + 0.5812(sp1.00d0.00)C7 in form A, which is not in form B. In form B there is an orbital σ(2)O3–C7 = 0.8605(sp1.00d0.00)O3 + 0.5095(sp1.00d0.00)C7,which is not in form A. These extra orbitals are of π character and indicate an increase in bond order. In form A, significant interactions of C6–C7, C8–C10 bond orbitals with antibonding orbitals of the O2–C5, O4–C27, C6–C7 σ2(C6–C7)→σ*2(O2–C5), σ2(C6–C7)→σ*2(O4–C27), σ2(C8–C10)→σ*2(C6–C7) with stabilization energies 34.18, 31.20, and 24.96 kcal/mol (Supplementary Information S4). Additionally, the molecule has lone electron pairs of oxygen atoms interactions with O2–C5, C5–C6, C6–C7, and O3–H44 bonds, n(LP2O1)→σ1*(O2–C5), n(LP2O2)→σ1*(O1–C5), n(LP2O2)→σ2*(C5–C6), and n(LP2O4)→σ1*(O3–H44) with energies 29.25, 40.10, 15.56, and 42.96 kcal/mol. In the tautomeric form B, the delocalization of electrons is maximal for the C8–C10 bond and is distributed over the antibonding orbitals O3–C7 σ2(C8–C10)→σ*2(O3–C7) with the stabilization energies 27.83 kcal/mol. Additionally, the molecule has lone electron pairs of oxygen atom interactions n(LP2O1)→σ2*(O2–C5), n(LP2O1)→σ1*(C8–C10), n(LP2O2)→σ1*(O1–C5), n(LP2O2)→σ1*(C5–C6), and n(LP2O3)→σ1*(O4–H44) with energies 31.36, 34.08, 37.96, 15.85, and 63.18 kcal/mol. 3.4. Vibrational Analysis The determination of the type of vibration in the experimental spectra was carried out by analyzing the potential energy, the atomic displacements, and the comparison with related compounds [29,30]. The experimental and calculated vibrational spectra for two tautomers A and B of compound 1 are shown in Figure 4 and Figure 5 and Table 3. The sharp medium-intensity peak in the IR spectrum at 3107 cm−1 and the frequency of 3108 cm−1 in the Raman spectrum refer to CH stretching vibrations (C8–H9 bond). Experimental frequencies in the region 3100–2960 cm−1 in vibrational spectra refer to νas(CH2) and νas(CH3) stretching vibrations. The symmetrical stretching vibrations of the methyl and methylene groups cause frequencies in the range of 2910–2860 cm−1 in the experimental spectra. Stretching of carbonyl groups without H-bonds (C5=O bond) causes a band at 1719 cm−1 in the experimental IR and Raman spectra (Figure 4 and Figure 5). The stretching vibrations of the carbonyl group forming the H-bond (C3=O7 bond) are shifted to the low-frequency region of 1636 cm−1. The bands in the region 1460–1400 cm−1 in the experimental spectra are due to the δas(CH3) and δas(CH2) bending vibrations. Symmetrical bending vibrations δs(CH3) and δs(CH2) cause bands in the region 1400–1340 cm−1. The frequency at 1315 cm−1 in the experimental IR spectrum and the band at 1318 cm−1 in the experimental Raman spectrum are due to the wagging vibrations of the methylene groups. Stretching vibrations of the CO and CC bonds of the pyran ring (C5–O1 and C5–C6 bonds) cause frequencies between 1290 and 1150 cm−1 in the experimental spectra. The bands in the region of 1060 to 1000 cm−1 in the experimental spectra were attributed to the deformation of the CCH angles and stretching of the CC bonds. The rocking vibrations of the methyl groups ρ(CH3)cause bands between 960 and 880 cm−1 in the experimental IR and Raman spectra. Bands of average intensity in the region of 860–760 cm−1 in the experimental spectra refer to CO and CC bonds (C5–O1 and C5–C6 bonds) stretching vibrations. Observed and calculated wavenumbers ν (cm−1), the intensity of the bands in the IR spectra I (km/mol) and relative intensity of the bands in the Raman spectra J (a.u.) and assignments for the tautomeric forms A and B of compound 1 in the gas phase by using the B3LYP/6–311++G** method. Abbreviations: vs, very strong; s, strong; m, medium; w, weak; vw, very weak; ν, stretching; β, deformation in plane; γ, deformation out of plane; wag, wagging; τ, torsion; βR, deformation ring; τR, torsion ring; ρ, rocking; τw, twisting; δ, deformation; a, antisymmetric; s, symmetric. The band at 713 cm−1 in the experimental spectra refers to the stretching vibrations of the CC bonds (C10–C11 bond). Bending vibrations of the pyran ring cause bands in the 620–470 cm−1 region in the experimental spectra. Bending and torsional vibrations of the pyran ring cause bands in the 500–100 cm−1 region of IR and Raman spectra. It is important to understand the changes that occur in the vibrational spectra of compound 1 during the tautomeric transformation. The spectra of tautomeric forms A and B are similar (Figure 4 and Figure 5). For tautomers A and B, the frequencies of most bands remain unchanged, but their intensity changes. Bands 1726, 1599, 1576, 1533, 1422, 1315, and 1192 cm−1 of the form A IR spectrum are shifted to frequencies 1721, 1617, 1580, 1536, 1400, 1341, and 1190 cm−1 of the form B IR spectrum (Figure 4). Bands 1726, 1599, 1576, 1533, 1433, 1340, 1316, 1278, 1249, 1198, 837, 686, 607, 578, 487 cm−1 in the Raman spectrum of form A are shifted to frequencies 1721, 1617, 1580, 1536, 1429, 1341, 1291, 1198, 853, 680, 579, 489 cm−1 in the Raman spectrum of form B (Figure 5). The theoretical spectra align with the experimental vibrational spectra of tautomer A across a wide frequency range (Figure 4 and Figure 5). Thus, the use of the DFT approximation for the considered molecular system is correct. 3.5. Hydrogen Bond Compound 1 has a strong H-bond with a cyclic chelate structure. The enol form of β-diketones is a well-known case of a 6-membered ring (chelate) with a strong resonance-assisted H-bond [31,32,33,34]. The characteristic spectral features observed for such structures include strong absorption bands in the region of 1580–1630 cm−1, instead of pronounced carbonyl stretching bands [16]. The stretching of the OH bond band shifts up to 2200 cm−1 [16]. Additionally, due to the resonance of two enol tautomers (both are chelate enol forms), this structural fragment can be considered a quasisymmetric O∙∙∙H∙∙∙O hydrogen bond. This bond has a broad and shallow potential well with two minima, which leads to a series of diffuse absorption bands scattered over a wide wavenumber region. The analysis of the observed IR spectrum 1 in the region of 2700–2000 cm−1 shows that there are several weak bands, ν(OH) (Figure 6). The calculated ν(OH) frequencies after scaling are 2682 and 2240 cm−1 for tautomers A and B, respectively, and they are in the range between 2700 and 2200 cm−1. An empirical formula is proposed that establishes a correlation between the frequencies ν(OH) observed and those calculated in the harmonic approximation: νobs = −757 + 1.173 νharm [33]. The calculation by this formula yields ν(OH)frequencies of 2520 and 1980 cm−1 for tautomers A and B, respectively. In the experimental IR spectrum of compound 1, the band 2524 cm−1 is observed, which can be attributed to ν(OH) vibrations. The shift of this band to low frequencies depends on the strength of the intramolecular H-bond. We see that in the most energy-stable tautomer A, the H-bond is weaker. This conclusion is consistent with the fact that the H-bond length for A and B tautomers is 2.473 and 2.418 Å, respectively. The strength of the H-bond can be described using Wiberg bond indices 0.158 (tautomer A) and 0.204 (tautomer B) [26]. These values of the Wiberg indices indicate that strong H-bonds are formed in compound 1 for both tautomers. It appears from our data that there is a correlation between the frequencies ν(OH)calculated in the harmonic approximation and the Wiberg indices. The stronger the H-bond, the lower the ν(OH) frequency and the higher the Wiberg index. The strength of the intramolecular H-bond in compound 1 can be estimated as the interaction n(LP2O4)→σ1*(O3–H44) with the energy 42.96 kcal/mol of the tautomer A and n(LP2O3)→σ1*(O4–H44) with energy 63.18 kcal/mol of tautomer B. The energy of the donor-acceptor interaction of the H-bond is larger for the B tautomer; therefore, it is stronger. Interactions σ1(O3–H44)→σ*1(C7–C8), σ2(C6–C7)→σ*2(O4–C27), σ2(C6–C7)→σ*2(C8–C10), σ2(C8–C10)→σ*2(C6–C7) with energies 5.83, 31.20, 7.49, 24.96 kcal/mol (tautomer A), and σ2(C8–C10)→σ*2(O3–C7) with energy 27.83 kcal/mol (tautomer B) are realized due to the conjugation of bonds in a six-membered ring.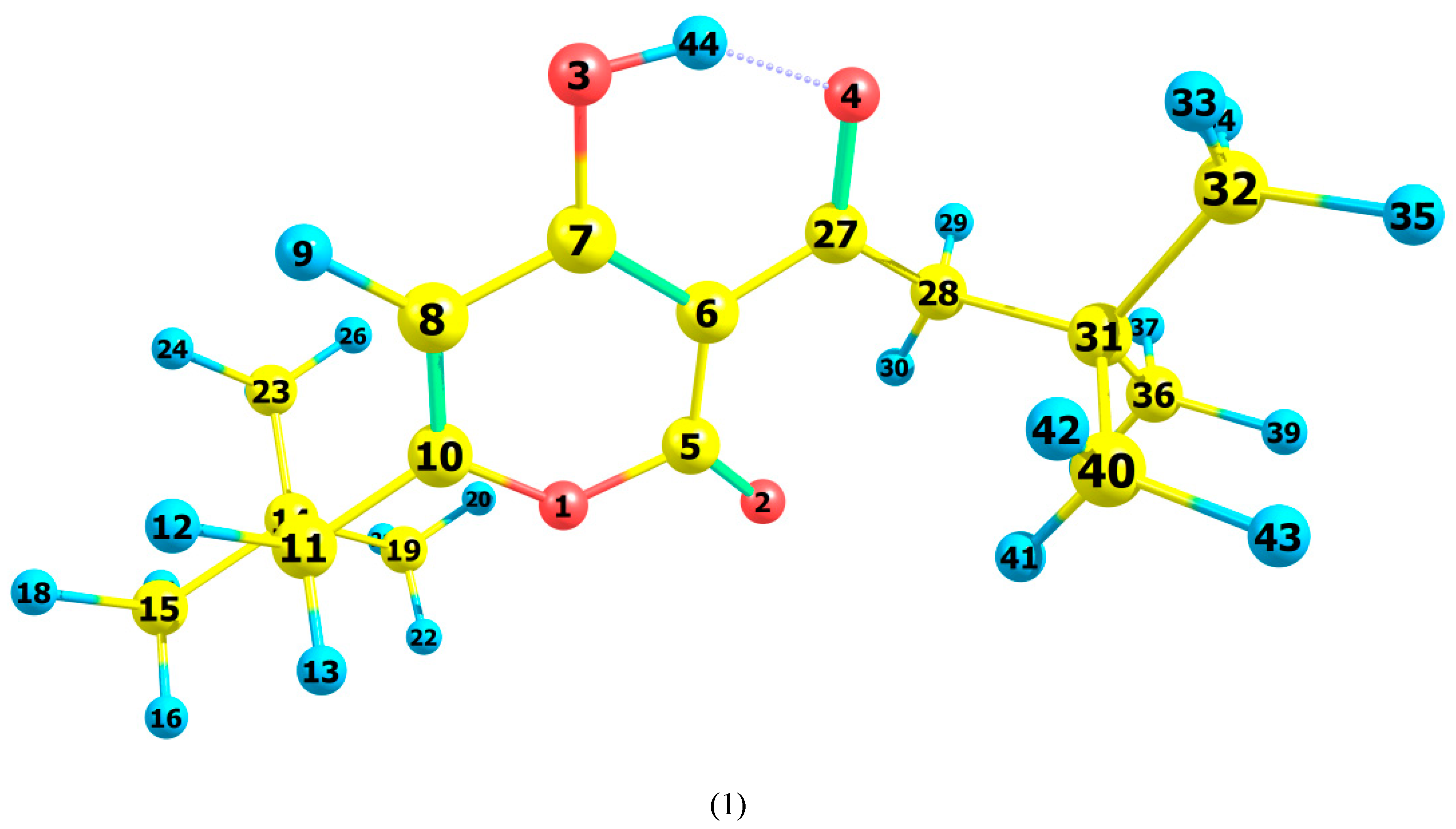 Table 1:
Table 1:
Gas
Chloroform
Dimethylsulfoxide
Tautomer
ΔG
p
ΔG
p
ΔG
p
A
0
90
0
87
0
90
B
1.35
10
1.12
13
1.32
10
Table 2:
Tautomer
I, eV
A, eV
|GAP|
μ, eV
S, eV
ω, eV
M, D
A
8.661
0.803
4.618
−4.732
0.127
2.850
3.414
B
8.795
0.883
4.693
−4.839
0.126
2.960
1.935
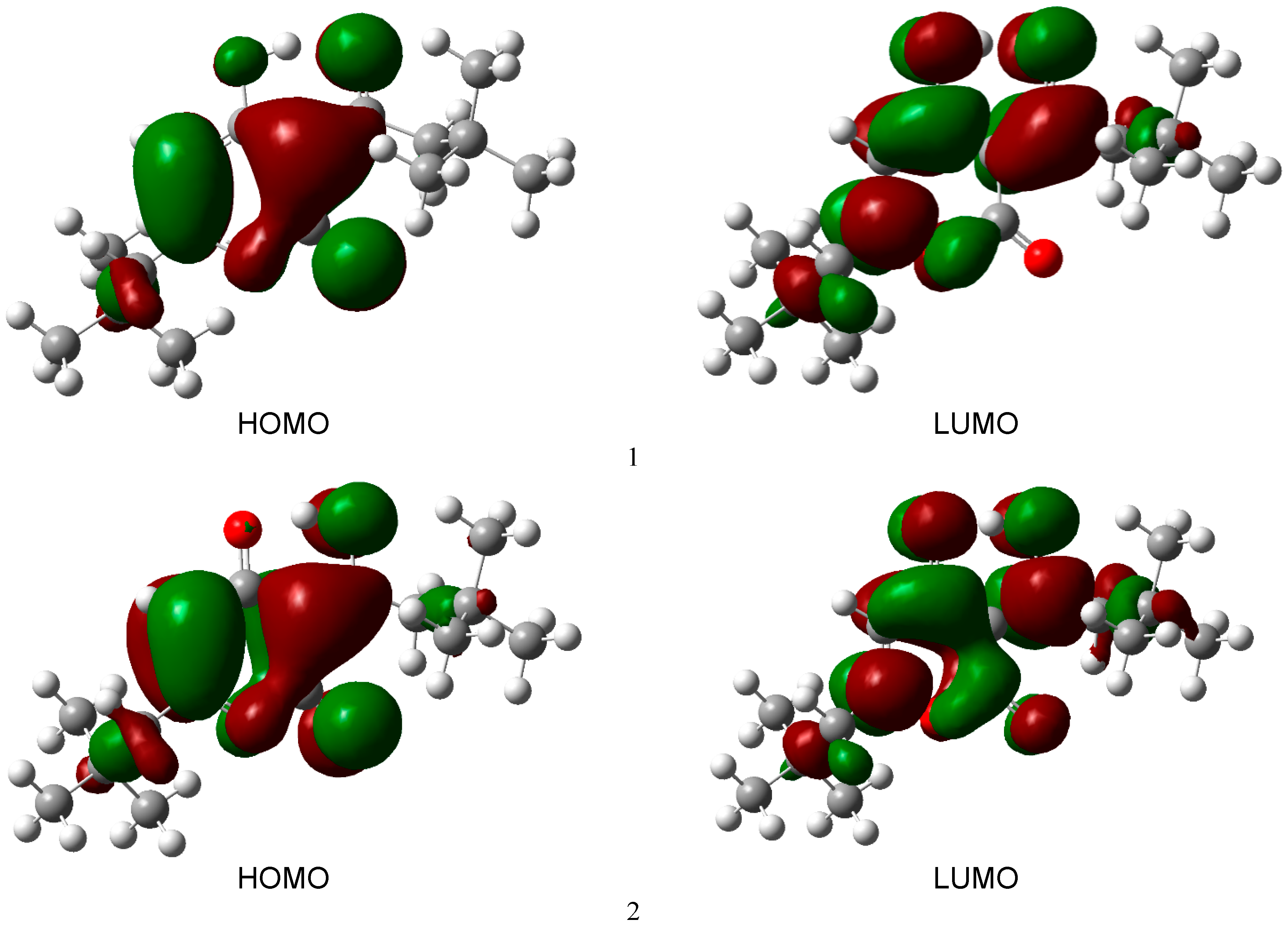
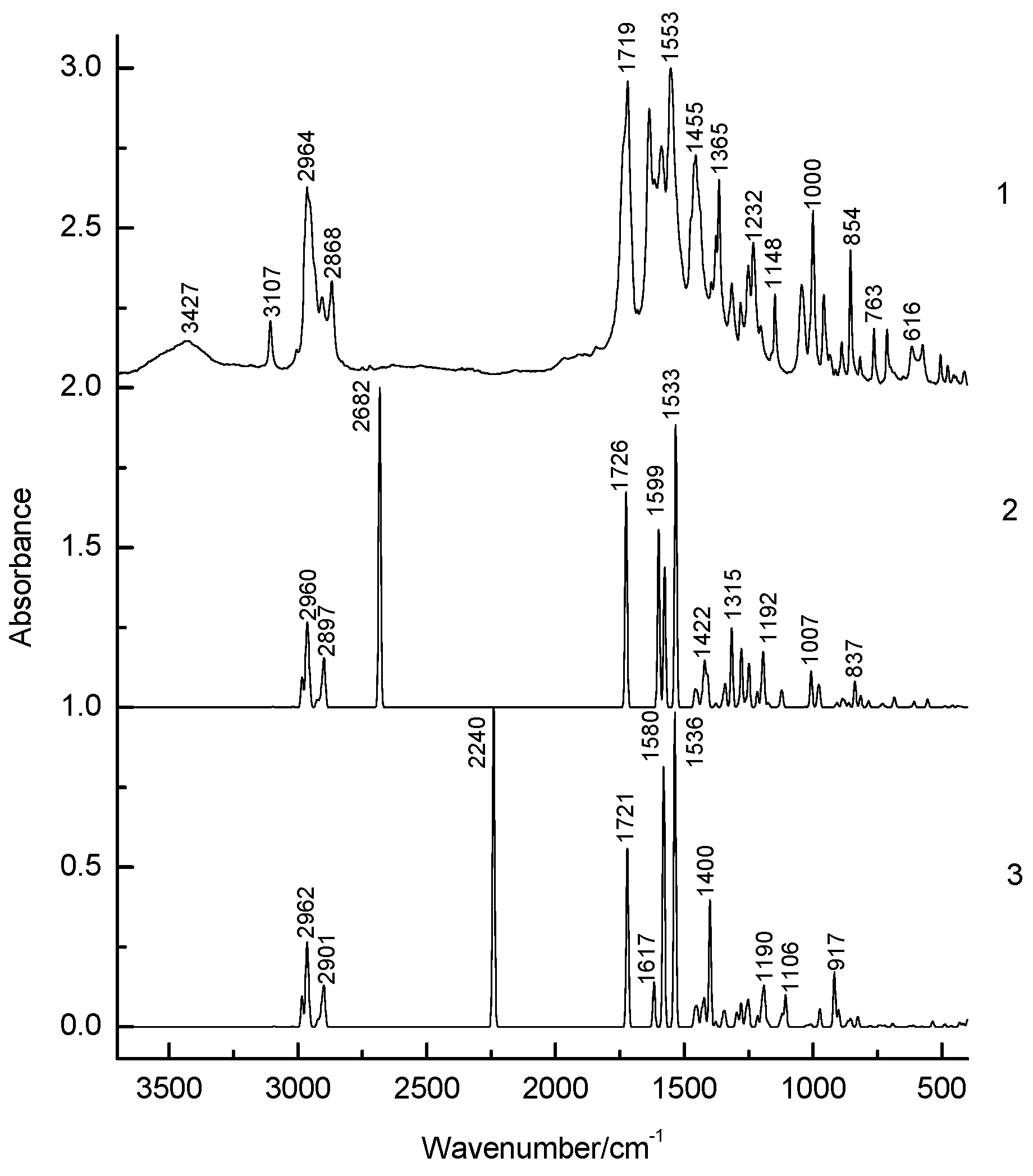
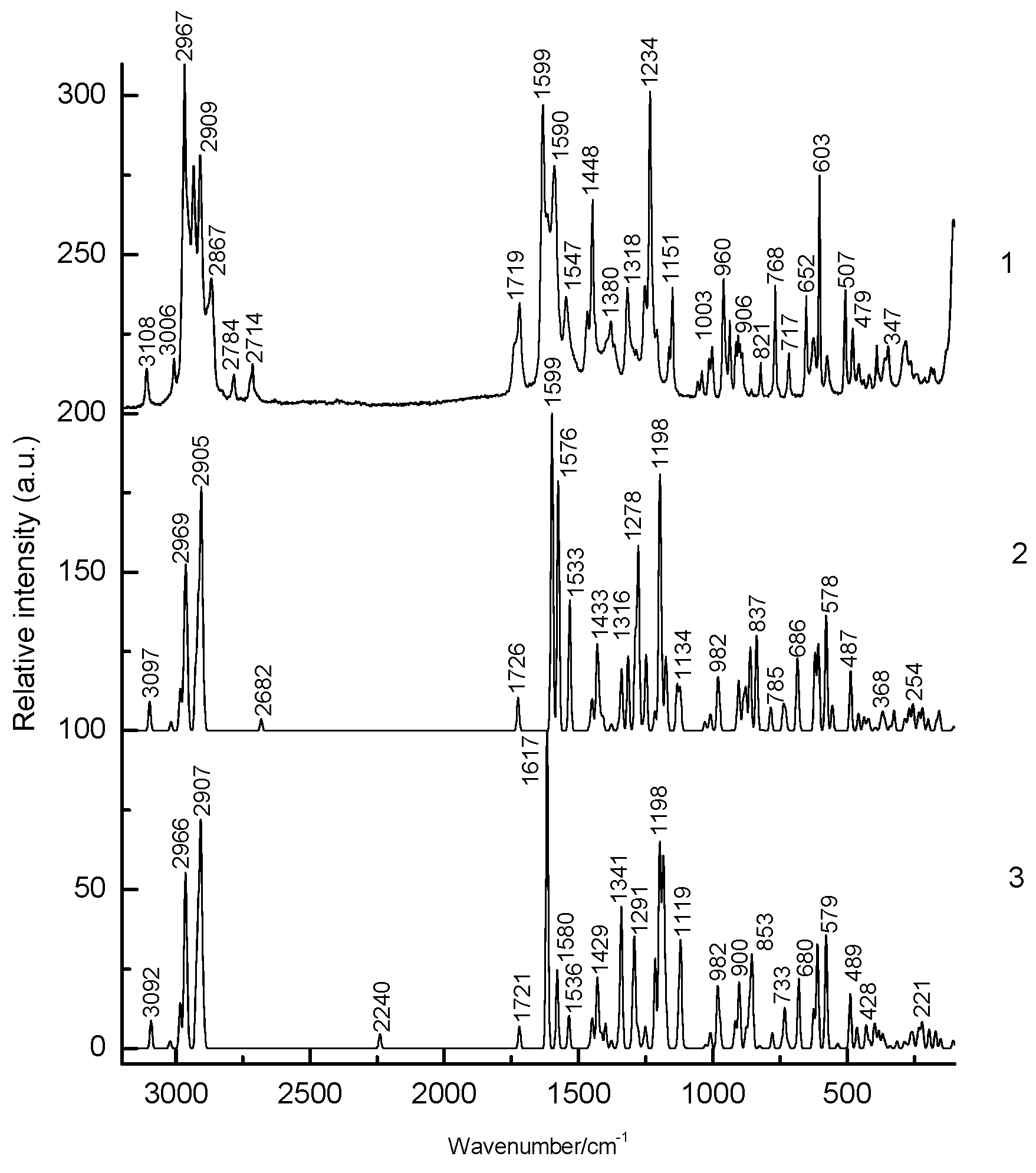 Table 3:
Table 3:
Experimental
B3LYP/6–311++G** Method
IR
Raman
A
B
Assignments
ν
ν
ν
I
J
ν
I
J
3427w
3107w
3108w
3097
1.1
9.2
3092
1.1
8.8
νC8–H9
3004vw
3006w
3018
1.0
2.7
3021
0.6
2.2
νC28–H29 νas CH2
2985
30.6
5.7
2984
26.8
5.5
νC19–H20 νas CH3
2984
26.3
4.7
2984
33.3
4.8
νC32–H33 νas CH3
2980
17.6
4.8
2983
16.2
4.2
νC40–H41 νas CH3
2969
64.4
14.8
2969
55.8
10.8
νC23–H24 νas CH3
2966
33.2
11.1
2966
43.1
13.4
νC15–H16 νas CH3
2964s
2967s
2964
43.8
12.8
2965
30.9
7.2
νC11–H12 νas CH2
2963
26.0
6.0
2963
45.5
12.3
νC36–H37 νas CH3
2961
3.1
2.1
2962
70.0
18.6
νC19–H21 νas CH3
2960
79.0
21.3
2961
1.9
1.8
νC32–H34 νas CH3
2956sh
2957
8.0
1.8
2957
6.7
1.8
νC23–H25 νas CH3
2956
3.8
1.5
2956
4.4
1.2
νC15–H17 νas CH3
2954
4.7
1.9
2956
22.0
2.7
νC32–H34 νas CH3
2953
26.1
2.9
2955
3.2
1.2
νC40–H42 νas CH3
2933m
2926
15.8
18.8
2923
14.8
20.7
νC28–H29 νas CH2
2916
14.1
37.1
2916
14.2
35.8
νC11–H12 νas CH2
2909m
2908
19.5
22.9
2908
20.3
21.8
νC19–H21 νs CH3
2906m
2905
26.0
50.4
2907
23.9
42.3
νC32–H33 νs CH3
2900
25.1
12.2
2901
26.5
1.4
νC23–H24 νs CH3
2899
28.5
1.0
2900
26.0
9.6
νC40–H41 νs CH3
2897
28.9
2.2
2896
28.7
3.4
νC15–H16 νs CH3
2868m
2867w
2895
31.1
4.6
2896
29.3
4.5
νC36–H37 νs CH3
2675vw
2784vw
2682
709.1
3.7
2240
787.7
4.4
νO3–H44
2666vw
2714vw
2666vw
2631vw
2608vw
2568vw
2524vw
2372vw
2364vw
2355vw
2338vw
1737sh
1738sh
1719vs
1719m
1726
479.7
10.4
1721
440.3
6.8
νC5=O2
1683w
1636s
1633vs
1599
394.4
100.0
1617
110.3
100.0
νC3=O7
1615w
1617sh
1588m
1590m
1576
311.0
78.6
1580
641.7
24.6
βO3–H44
1553vs
1547m
1533
626.8
41.2
1536
775.5
10.3
νC6–C7
1474sh
1462sh
1467m
1460
18.0
0.7
1460
21.1
0.4
βC32–H33 δas CH3
1455m
1458
17.0
0.9
1458
19.6
0.6
βC19–H20 δas CH3
1452
12.7
1.5
1451
19.4
1.6
βC36–H39 δas CH3
1451
4.9
5.1
1450
14.0
3.7
βC15–H16 δas CH3
1449
6.7
1.5
1449
6.6
1.4
βC11–H12 δas CH2
1445sh
1448m
1447
11.3
2.6
1447
10.0
3.4
βC28–H29 δas CH2
1437sh
1438sh
1433
7.8
11.7
1432
20.9
5.3
βC40–H41 δas CH3
1430
8.7
6.0
1432
15.3
6.1
βC32–H33 δas CH3
1429
3.4
7.4
1429
2.0
7.5
βC15–H16 δas CH3
1428
6.4
4.2
1428
3.5
5.0
βC19–H20 δas CH3
1425
0.4
0.2
1425
0.4
0.2
βC23–H25 δas CH3
1424
2.4
0.3
1424
2.0
0.0
βC36–H37 δas CH3
1422
77.7
4.5
1422
63.1
1.3
βC28–H29 δs CH2
1417
32.8
2.2
1415
6.4
4.6
βC11–H12 δs CH2
1394vw
1409
64.0
4.0
1400
312.5
7.8
βC28–H29 δs CH2
1376w
1380m
1379
4.3
1.4
1379
5.8
1.1
βC19–H21 δs CH3
1365m
1369sh
1376
4.7
0.8
1377
6.8
1.3
βC32–H35 δs CH3
1365sh
1351
7.6
1.7
1351
6.2
3.7
βC19–H20 δs CH3
1349
6.0
0.3
1350
14.3
0.3
βC32–H33 δs CH3
1347
7.3
0.3
1346
8.3
1.2
βC23–H24 δs CH3
1344
7.5
0.6
1345
8.6
0.6
βC36–H37 δs CH3
1340
44.1
18.9
1341
23.0
43.3
νC5–O1
1315w
1318m
1316
176.4
23.6
1298
30.1
13.8
βC28–H29 wag CH2
1289
8.9
29.5
1291
13.8
29.5
βC11–H12 wag CH2
1282w
1285vw
1278
129.0
57.2
1279
58.2
5.9
νC6–C7
1261
16.8
2.1
1260
39.7
1.7
βC28–H29 wag CH2
1251w
1253m
1249
97.4
23.9
1251
63.1
6.6
νC6–C27
1232m
1234vs
1216
34.5
6.1
1215
26.9
28.2
νC6–C27
1203w
1208w
1198
57.3
72.3
1198
60.9
61.8
βC23–H24 wag CH3
1192
98.5
21.7
1190
90.7
22.0
βC32–H35 wag CH3
1177
4.7
10.8
1184
8.2
49.9
νC5–C6
1174
2.9
12.4
1176
7.5
3.0
βC23–H24 τw CH3
1161sh
1163w
1170
4.2
3.2
1174
3.4
9.7
νC5–O1
1148m
1151m
1134
2.0
14.2
1128
11.0
8.1
βC8–H9
1123
25.1
11.1
1122
3.4
16.2
βC28–H29 τw CH2
1056vw
1119
19.1
3.6
1119
29.0
18.1
βC11–H12 τw CH2
1044m
1041w
1030
0.7
2.7
1106
78.5
0.4
βC40–H41
1014w
1017
0.6
0.8
1027
2.2
1.0
βC23–H24
1011
1.2
1.7
1018
4.0
0.3
βC36–H39
1009
15.2
2.4
1010
1.7
2.7
βC23–H24 τw CH3
1000m
1003w
1007
64.9
1.3
1008
4.1
2.3
δO3–H44
982
22.5
13.9
982
1.2
18.8
νC7–C8
959m
960m
976
41.1
7.2
973
44.3
7.2
νO1–C10
935w
937w
928
0.1
0.1
928
0.5
0.1
γC32–H33 ρ CH3
929sh
926
0.0
0.1
926
0.2
0.1
γC23–H24 ρ CH3
912vw
914w
910
3.7
2.7
917
135.1
8.4
γC28–H29 ρ CH3
906w
907
4.9
2.2
907
1.4
1.3
γC19–H20 ρ CH3
903
2.1
8.9
903
2.9
7.7
νC14–C23
899w
902
0.4
4.4
902
3.7
3.1
νC31–C36
888w
891w
887
17.5
9.3
900
35.3
10.5
νC5–C6, νC5–O1
877
13.4
12.9
876
1.0
6.3
νC14–C19
861sh
862
3.2
3.9
865
12.8
11.0
νC14–C15
854m
856vw
861
6.9
22.6
857
5.1
14.3
νC10–C11
844sh
837
57.4
30.0
853
16.6
18.5
νO1–C5
817w
821w
815
26.3
0.3
826
25.0
0.7
γC8–H9
779sh
768m
785
13.7
7.4
779
2.5
4.8
ρC27–O4
763w
754sh
738
2.6
7.6
742
4.5
2.4
νC8–C10
729
7.2
5.7
733
2.1
12.5
ρC7–O3
713w
717w
720
1.4
0.2
721
2.8
1.9
νC10–C11
697sh
686
18.3
21.3
691
7.4
0.6
νC27–C28
686sh
652m
680
6.8
3.9
680
0.3
21.8
ρC7–O3
650sh
625w
620
1.1
24.4
625
2.2
12.2
νC10–O1
616w
603m
607
12.3
27.0
611
2.8
32.7
ρC7–O3
574w
575w
578
0.4
36.4
579
0.8
35.6
νC10–C11
544sh
555
17.8
8.0
535
12.8
1.5
βR
536sh
529sh
505w
507m
477vw
479w
487
2.6
18.8
489
6.0
17.1
βR
453vw
456w
458
4.1
5.4
464
2.0
6.6
βR
445vw
438
3.1
4.3
430
10.3
2.6
βR
436vs
439vw
426
1.7
1.9
428
0.0
4.8
βR
412vw
418vw
419
0.2
3.1
417
8.3
2.4
τR
396
0.1
0.7
400
14.6
5.2
τR
389w
391
3.1
0.5
395
4.8
3.9
βC27–O4
381sh
375
3.5
2.4
383
11.1
5.5
ρC5=O2
368
0.3
5.3
369
0.4
4.4
βC14–С19
359sh
359
0.6
2.9
360
3.0
1.8
βC27–С28
347w
340
2.1
1.6
340
0.9
0.8
γC10–С11
326
6.6
6.4
315
3.0
2.1
βC27–O4
288sh
287
0.4
3.0
288
0.2
1.7
τC14–С15
281w
283
1.5
0.6
283
0.2
0.3
τC31–С32
280
0.3
0.4
280
0.2
0.2
τC14–С15
276
0.1
1.1
277
0.1
0.9
τC14–С23
264vw
269
2.2
6.5
265
2.1
2.8
τR
258
1.0
2.6
260
0.3
2.2
τC14–С23
254
0.9
6.5
255
0.5
3.5
βR
241vw
235
1.3
4.5
236
0.4
4.3
τC14–С23
230
0.2
1.9
230
1.5
3.7
τC31–С40
220
0.6
6.0
221
0.3
6.1
τC14–С15
211vw
219
0.3
1.1
219
0.6
2.0
τR
188vw
199
3.6
3.6
195
0.7
5.9
τR
178vw
167
0.3
3.1
171
1.0
5.4
τR
168vw
157
2.7
6.2
152
2.5
3.0
τR
105vw
103
0.2
1.3
105
0.9
2.4
τR
In conclusion, the correlation between the structure and H-bonding was established in 3-(3,3-Dimethylbutanoyl)-4-hydroxy-6-neopentyl-2H-pyran-2-one. For pyrone 1, the X-ray diffraction, DFT-calculations and IR, Raman spectroscopy revealed the most favourable tautomeric form A. The content of tautomer B increases in the nonpolar solvent but does not exceed 13%. As can be seen from the calculations and experimental X-ray data, the pyran ring of the molecule is flat. A satisfactory agreement is observed between the calculated geometrical parameters of tautomer A and the experimental X-ray data. The calculation of the normal vibrations by the DFT method gives a detailed description of the dynamics of pyrone 1. The intensities of the bands in the IR spectra show high sensitivity to the H-bond in compound 1. The HOMO and LUMO orbitals of the acid molecule are located on the pyran ring. During tautomeric transformations, there is a significant delocalization of the charge, which modifies the reactivity of the molecule. The reactivity of compound 1 was characterized using descriptors. Form B was found to have higher ionization energy, electron affinity, chemical potential, and electrophilic index than Form A. The dipole moment is higher for Form A, and the softness of the two molecules is the same. The obtained results provide the opportunity better understand the interplay between the tautomeric flexibility of the pyrone ring and its H-bonding, providing a new approach for rational design of drugs with desired properties.
| IR | Infrared |
| DFT | Density Functional Theory |
| NMR | Nuclear Magnetic Resonance |
| FTIR | Fourier Transform Infrared |
| NBO | Natural Bond Orbital |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
| IE | Ionization Energy |
| EA | Electron Affinity |
| FWHM | Full Width at Half Maximum |
V.F.: conceptualization, methodology, software, writing-original draft preparation and editing. A.V.: investigation of IR and Raman spectra. E.N., M.M.: synthesis of pyran. V.K.: conceptualization, methodology, reviewing and editing. V.K.: conceptualization, methodology, reviewing and editing.
No datasets were generated or analyzed during the current study.
Not applicable.
The authors declare no competing interests.
The work was carried out within the framework of the state assignment “Petrochemistry and Catalysis. Rational use of carbon-containing raw materials”, No. 121031300092-6.
The authors are grateful to the Assigned Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for technical assistance in research.
Supplementary materials are available for download here.
[1] Penta, S. . Dehydroacetic Acid and Its Derivatives: Useful Synthons in Organic Synthesis ; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; . .
[2] Dobler, D.; Leitner, M.; Moor, N.; Reiser, O. 2-Pyrone—A privileged heterocycle and widespread motif in nature. Eur. J. Org. Chem. 2021, 2021, 61801–6205. [CrossRef]
[3] Tian, R.D.; Sheng, M.K.; Zhao, Y.F.; Wen, C.N.; Liu, M.; Ma, J. α-Pyrone derivatives from the endophytic Fusarium sp. L33 isolated from Dioscorea opposita. Phytochem. Lett. 2024, 62, 14–17. [CrossRef]
[4] Colin, M.J.; Aguilar, M.A.; Martin, M.E. A theoretical study of solvent effects on the structure and UV-vis spectroscopy of 3-hydroxyflavone (3-HF) and some simplified molecular models. ACS Omega 2023, 8, 19939–19949. [CrossRef] [PubMed]
[5] Fujihara, K.; Hashimoto, T.; Sasaki, H.; Koyama, K.; Kinoshita, K. Inhibition of Aβ aggregation by naphto-γ-pyrone derivatives from a marine-derived fungus, Aspergillus sp. MPUC239. J. Nat. Med. 2023, 77, 516–522. [CrossRef] [PubMed]
[6] Kawsar, S.M.A.; Hosen, M.A.; Chowdhury, T.S.; Rana, K.M.; Fujii, Y.; Ozeki, Y. Thermochemical, PASS, molecular docking, drug-likeness and in silico admet prediction of cytidine derivatives against HIV-1 reverse transcriptase. Rev. Chim. Lett. 2023, 72, 159–178. [CrossRef]
[7] Tabassum, R.; Kawsar, S.M.A.; Alam, A.; Saha, S.; Hosen, A.; Hasan, I.; Prinsa; Mohhamed, C. Synthesis, spectral characterization, biological, FMO, MEP, molecular docking, and molecular dynamic simulation studies of cytidine derivatives as antimicrobial and anticancer agents. Chem. Phys. Impact 2024, 9, 100724. [CrossRef]
[8] Akter, N.; Bourougaa, L.; Oussaf, M.; Bhowmic, R.C.; Uddin, K.M.; Bhat, A.R.; Ahmed, S.; Kawsar, S.M.A. Molecular docking, ADME-Tox, DFT and molecular dynamic simulation of butyroyl glucopyranoside derivatives against DNA gyrase inhibitors as antimicrobial agents. J. Mol. Struct. 2024, 1307, 137930. [CrossRef]
[9] Guo, Z.; Chen, B.; Chen, D.; Deng, X.; Yuan, J.; Zhang, S.; Xiong, Z.; Xu, J. New isocoumarin and pyrone derivatives from the Chinese Mangrove plant Rhizophora mangle-associated fungus Phomopsis sp. DHS-11. Molecules 2023, 28. [CrossRef] [PubMed]
[10] Jiang, W.; Hou, W.; Yan, C.; Nie, Z.; Chang, Q.; Li, X.; Liu, W. Synthesis and performance of deep-red phosphorescent iridium complexes with pyrone as an auxiliary ligand. Molecules 2024, 29. [CrossRef]
[11] Kohanov, Z.A.; Shuvo, S.I.; Lowell, A.N. Regioselective annulations of 6-carboxy-substituted pyrones asa two-carbon unit in formal [4 + 2]cycloaddition reactions. J. Org. Chem. 2024, 89, 9557–9568. [CrossRef] [PubMed]
[12] Chalaca, M.Z.; Figuerola-Villar, J.D. A theoretical and NMR study of the tautomerism of dehydroacetic acid. J. Mol. Struct. 2000, 554, 225–231. [CrossRef]
[13] Gorodetsky, M.; Luz, Z.; Mazur, Y. Oxygen-17 nuclear magnetic resonance studies of the equilibria between the enol forms of β-diketones. J. Am. Chem. Soc. 1967, 89, 1183–1189. [CrossRef]
[14] Billes, F.; Eleckova, L.; Mikosch, H.; Andruch, V. Vibrational spectroscopic study of dehydroacetic acid and its cinnamoyl pyrone derivatives. Spectrochim. Acta Part A 2015, 146, 97–112. [CrossRef] [PubMed]
[15] Emsley, J. The composition, structure and hydrogen bonding of the β-diketones. Complex Chem. 1984, 57, 147–191. [CrossRef]
[16] Yamada, K. Infrared and ultraviolet spectra of α- and γ-pyrones. Bull. Chem. Soc. Jpn. 1962, 35, 1323–1329. [CrossRef]
[17] Tykhanov, D.A.; Serikova, I.I.; Yaremenko, F.G.; Roshal, A.D. Structure and spectral properties of cinnamoyl pyrones and theirs vinylogs. Cent. Eur. J. Chem. 2010, 8, 347–355. [CrossRef]
[18] Nomerotskaya, E.I. Polyfunctional alpha-Pyrones: Direct Synthesis from Carboxylic Acids and Properties. Unpublished Master’s Thesis, Moscow State University, Moscow, Russia, 2020. .
[19] Sun, X.; Gong, M.; Huang, M.; Li, Y.; Kim, J.K.; Kovalev, V.V.; Shokova, E.A.; Wu, Y. One-pot synthesis of pyrones from aromatic ketones/heteroarenes and carboxylic acids. J. Org. Chem. 2020, 85, 15051–15061. [CrossRef]
[20] . Available online: https://plastics-polymer-analysis.com/ru/ (accessed on 1 January 2020).
[21] Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [CrossRef]
[22] Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [CrossRef] [PubMed]
[23] Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. . Gaussian 09 Revision C.01 ; Gaussian Inc.: Wallingford, CT, USA, 2010; . .
[24] Jamroz, M.H. Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim. Acta 2013, 114, 220–230. [CrossRef] [PubMed]
[25] Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. Comput. Mol. Sci. 2012, 2, 1–42. [CrossRef]
[26] Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [CrossRef]
[27] Tomasi, J.; Perisco, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [CrossRef]
[28] Castillo, M.V.; Rudyk, R.A.; Davies, L.; Brandan, S.A. Analysis of the structure and the FT-IR and Raman spectra of 2-(4-nitrophenyl)-4H-3,1-benzoaxin-4-one. Comparison with the chlorinated and methylated derivatives. J. Mol. Struct. 2017, 1140, 2–11. [CrossRef]
[29] Chahar, F.C.; Alvarez, P.A.; Zampini, C.; Isla, M.I.; Brandan, S.A. Experimental and DFT studies on 2’,4’-dihydroxychalcone, a product isolated from Zuccagnia punctata Cav. (Fabaceae) medicinal plant. J. Mol. Struct 2020, 1201, 127221. [CrossRef]
[30] Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [CrossRef]
[31] Rusinska-Roszak, D. Intramolecular O-H∙∙∙C=O hydrogen bond energy via the molecular tailoring approach to RAHB structures. J. Phys. Chem. A 2015, 119, 3674–3687. [CrossRef]
[32] Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Pendas, A.M.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [CrossRef]
[33] Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of strong intramolecular hydrogen bonds. Molecules 2017, 22. [CrossRef]
[34] Wiberg, K.A. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [CrossRef]