
 ACCESS
Review Article
ACCESS
Review Article
Volume 1, Article ID: 2025.0004

Nosayba Al-Damook
nosayba.aldamook@aau.ac.ae


Molham Sakkal
mohammad.sakkal@aau.ac.ae


Sedra Kremesh
sedra.kremish@gmail.com

Walaa K. Mousa
walaa.mousa@aau.ac.ae

Mostafa Khair
mrk6@nyu.edu


Ghalia Khoder
gkhoder@sharjah.ac.ae

Rose Ghemrawi
rose.ghemrawi@aau.ac.ae
1 College of Pharmacy, Al Ain University, Abu Dhabi P.O. Box 112612, United Arab Emirates
2 AAU Health and Biomedical Research Center, Al Ain University, Abu Dhabi P.O. Box 112612, United Arab Emirates
3 College of Pharmacy, Mansoura University, Mansoura 35516, Egypt
4 Core Technology Platforms, New York University Abu Dhabi, Abu Dhabi P.O. Box 129188, United Arab Emirates
5 Department of Pharmaceutics and Pharmaceuticals Technology, College of Pharmacy, University of Sharjah, Sharjah 27272, United Arab Emirates
6 Sharjah Institute for Medical Research, University of Sharjah, Sharjah 27272, United Arab Emirates
* Author to whom correspondence should be addressed
Received: 30 Jan 2025 Accepted: 11 Apr 2025 Available Online: 12 Apr 2025 Published: 28 Apr 2025
Cancer cells sustain rapid growth by reprogramming their metabolism to depend heavily on glycolysis, enhanced insulin signaling, and adaptive changes in the unfolded protein response (UPR). Insulin Potentiation Therapy (IPT) enhances the efficacy of chemotherapy by increasing insulin receptor expression, thereby improving the uptake of chemotherapeutic agents. Meanwhile, targeting endoplasmic reticulum (ER) stress disrupts the protein-folding machinery within cancer cells, steering them toward apoptosis. However, IPT faces several limitations, including limited clinical evidence, variable responses across tumor types, and concerns regarding safety and standardization. This review is the first to comprehensively examine the combined modulation of ER stress and IPT, highlighting novel clinical applications and presenting supporting evidence for this synergistic approach in cancer treatment. We compare the distinct metabolic features of cancer cells with those of normal cells and analyze how these differences contribute to tumor progression, the critical role of ER stress in cancer development, and the influence of insulin on cancer metabolism. In addition, we explore the potential synergy between IPT and ER stress modulation by delving into the mechanisms underlying IPT, the benefits of integrating ER stress modulation with IPT, and how insulin can modulate ER stress to improve therapeutic outcomes.
To ensure continuous and rapid growth, cancer cells reprogram their metabolism by shifting from mitochondrial oxidative phosphorylation to aerobic glycolysis—even in the presence of oxygen. This phenomenon, known as the “Warburg effect”, is illustrated in Figure 1. This metabolic switch prioritizes the production of biosynthetic intermediates over efficient ATP generation, enabling cancer cells to accumulate the nucleotides, amino acids, and lipids essential for rapid cell division [1]. Many cancer cells are heavily dependent on glutamine, which serves as an important source of reduced nitrogen for biosynthetic reactions and as a source of carbon to replenish the tricarboxylic acid (TCA) cycle, produce glutathione, and provide precursors for nucleotide and lipid synthesis [2,3]. Unlike normal cells, cancer cells rely heavily on metabolic pathways such as aerobic glycolysis and glutamine metabolism to support their rapid growth and biosynthetic demands. This metabolic dependency creates therapeutic opportunities, as targeting key enzymes in these pathways can selectively impair cancer cell survival while minimizing harm to normal tissues [3]. For example, pharmacological inhibitors of glycolytic enzymes or glutamine metabolism can selectively disrupt cancer cell viability while sparing normal cells, demonstrating promising potential in both preclinical and clinical studies [4,5]. The endoplasmic reticulum (ER) plays a central role in synthesizing, folding, and modifying secreted and transmembrane proteins. Disruptions to its protein-folding capacity, caused by external factors or intrinsic cellular events, lead to the accumulation of unfolded or misfolded proteins—a condition termed ER stress. Activation of the unfolded protein response (UPR) occurs when misfolded proteins accumulate in the endoplasmic reticulum (ER), prompting the cell to restore homeostasis by upregulating genes that encode ER chaperones and components of ER-associated degradation (ERAD) [6,7]. In cancer cells, persistent ER stress is common due to genetic, transcriptional, and metabolic abnormalities [8,9,10,11]. The UPR is mediated by three key ER membrane-localized sensors: inositol-requiring enzyme 1α (IRE1α), eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3, also known as PERK), and activating transcription factor 6 (ATF6). Under normal conditions, these sensors are maintained in an inactive state through binding with immunoglobulin-binding protein (BiP, also known as GRP78). Accumulation of unfolded or misfolded proteins triggers BiP’s release from IRE1α, PERK, and ATF6, activating their respective signaling pathways [12]. Activation of the unfolded protein response (UPR) occurs when misfolded proteins accumulate in the endoplasmic reticulum (ER), prompting the cell to restore homeostasis by upregulating genes that encode ER chaperones and components of ER-associated degradation (ERAD) [6,7]. In cancer cells, persistent ER stress is common due to genetic, transcriptional, and metabolic abnormalities [8,9,10,11]. The UPR is mediated by three key ER membrane-localized sensors: inositol-requiring enzyme 1α (IRE1α), eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3, also known as PERK), and activating transcription factor 6 (ATF6). Under normal conditions, these sensors are maintained in an inactive state through binding with immunoglobulin-binding protein (BiP, also known as GRP78). Accumulation of unfolded or misfolded proteins triggers BiP’s release from IRE1α, PERK, and ATF6, activating their respective signaling pathways [12]. Among these branches, the IRE1-XBP1 pathway is the most conserved. During UPR activation, IRE1 oligomerizes and auto-phosphorylates its kinase domain, initiating its RNase activity. This activity splices XBP1 mRNA, excising 26 nucleotides to produce a potent transcription factor that enhances protein folding, ERAD, and lipid biosynthesis [13,14]. IRE1 also regulates mRNA degradation through regulated IRE1-dependent decay (RIDD) and activates JNK signaling, mediating cytoprotective responses [13]. PERK, another UPR branch, phosphorylates eIF2α at Ser51, transiently inhibiting global protein synthesis while selectively enhancing ATF4 translation. ATF4 regulates genes involved in autophagy, ERAD, and apoptosis [15,16]. To restore protein synthesis equilibrium, eIF2α is dephosphorylated by a GADD34-PP1 complex, dynamically regulating protein synthesis during ER stress [17]. ATF6, a type II transmembrane protein, is activated in the Golgi after cleavage by S1P and S2P proteases. The active ATF6 regulates targets like BiP/GRP78, GRP94, and CHOP, and enhances ERAD in partnership with XBP1 [18]. The metabolic vulnerabilities created by ER stress and UPR activation provide a unique opportunity to synergize with therapies like IPT. By leveraging the reliance of cancer cells on insulin and IGF signaling pathways, IPT combines insulin with chemotherapeutic agents to enhance selective drug uptake, potentially reducing systemic toxicity and improving therapeutic outcomes. However, targeting metabolic pathways and ER stress responses in cancer therapy poses challenges, including potential toxicity to normal cells, tumor heterogeneity, and the risk of adaptive resistance mechanisms [4,19]. Despite the available literature on IPT and its potential to enhance chemotherapy efficacy, its potential synergy with ER stress modulation remains largely unexplored. This review aims to bridge this gap by evaluating the interplay between IPT and ER stress pathways, providing a novel perspective on their combined therapeutic potential in cancer treatment. IPT remains underexplored and requires further research to standardize its mechanisms and clinical efficacy [20,21].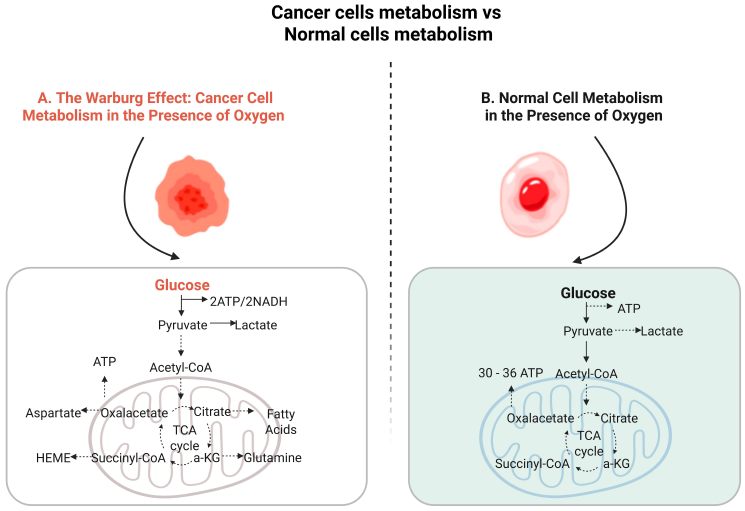
Impaired ER proteostasis is a hallmark of cancer, driven by factors in the tumor microenvironment (TME) such as hypoxia, reactive oxygen species (ROS), and metabolic imbalances, which trigger ER stress and activate the UPR [22,23]. Cancer cells exploit heightened UPR activation to survive ER stress-induced damage, promoting tumor progression and resistance to chemotherapy [23]. The three major UPR branches—IRE1α, PERK, and ATF6—play pivotal roles in mediating cancer cell survival and regulating apoptosis [24]. Elevated IRE1α signaling supports tumor growth and immune evasion by suppressing anti-tumor immunity. Pharmacological inhibition of IRE1α has demonstrated promise in reducing tumor growth and enhancing immunotherapy responses [25]. Similarly, GRP78, a central UPR regulator, has been shown to prevent apoptosis and contribute to chemotherapy resistance. Targeting GRP78 induces apoptosis and sensitizes cancer cells to chemotherapeutic agents [26]. Bioinformatics and survival analyses have identified high ATF6 expression as a poor prognostic marker in cancer, correlating with unfavorable survival outcomes [27]. In vitro studies further highlight ATF6’s oncogenic role; silencing ATF6 in oral squamous cell carcinoma cells suppresses proliferation and migration while promoting autophagy and apoptosis, underscoring its importance in tumor progression [27]. PERK-mediated UPR activation also enhances cancer cell survival. In melanoma models, PERK inhibition induces immunogenic cell death (ICD) and paraptosis, thereby enhancing anti-tumor immune responses [28]. The dual role of the UPR in regulating apoptosis is a key factor in cancer progression. Early UPR activation promotes cell survival, but prolonged ER stress often triggers apoptotic pathways. For instance, inhibiting the IRE1/XBP-1s pathway with the prodrug TC-D-F07 induces apoptosis in tumor cells [29]. Curcumin-induced apoptosis in colorectal cancer through ATF6 signaling further exemplifies the therapeutic potential of modulating UPR pathways [30]. Additionally, the cardiac glycoside oleandrin induces apoptosis in breast cancer cells via the PERK/eIF2α/ATF4/CHOP pathway, linking ER stress modulation to enhanced pro-apoptotic activity [31]. The UPR also contributes to chemotherapy resistance by helping cancer cells adapt to therapeutic stress. In triple-negative breast cancer, UPR activation through IRE1 and PERK reduces doxorubicin (Dox) internalization, activates Nrf2, and downregulates PDCD4, thereby disrupting protein translation and promoting chemoresistance. Co-treatment with cryptotanshinone restores Dox sensitivity [32]. Similarly, in pancreatic ductal adenocarcinoma (PDAC), RUNX1 upregulates the BiP/PERK/eIF2α pathway, enhancing stress adaptation and resistance to gemcitabine (GEM). RUNX1 inhibition improves GEM efficacy by reducing resistance and promoting apoptosis [33]. Chronic ER stress plays a crucial role in shaping the TME and facilitating immune evasion. Persistent UPR activation in cancer and immune cells promotes tumor progression through mechanisms such as XBP1-regulated cholesterol synthesis. Cholesterol secreted in extracellular vesicles is internalized by myeloid-derived suppressor cells (MDSCs), enhancing their immunosuppressive functions and weakening anti-tumor immune responses [34]. Additionally, hypoxic conditions in the TME activate HIF-2α, which enhances stemness and chemoresistance in breast cancer cells by upregulating SOD2, reducing mitochondrial ROS, and altering protein disulfide isomerase (PDI) activity. These changes further promote chemoresistance and the acquisition of stem-like properties [35].
Cancer cells are characterized by their unusual reliance on aerobic glycolysis to meet their energy requirements rather than mitochondrial oxidative phosphorylation as in regular cells [36]. This metabolic reprogramming is responsible for the rapid growth rate of malignant cells in comparison to normal cells [37]. Although aerobic glycolysis under the hypoxic tumor microenvironment of the cancer cells generates a much smaller amount of ATP compared with aerobic respiration, the excessive expression of GLUT in tumor cells enables these cells to meet their energy needs through maximizing glucose uptake, to provide sufficient raw materials for aerobic glycolysis and ATP production [37]. Insulin is essential for activating multiple cellular homeostasis pathways in normal cells; however, in cancer cells, insulin receptors are often overexpressed, and insulin signaling pathways are dysregulated [38]. Insulin is an anabolic hormone recognized as a modulator of cancer metabolism, which has been associated with the development of various malignancies, including breast [39], colon [40], and lung cancer [41,42]. Insulin facilitates the uptake of essential building blocks like glucose, potassium, phosphate, and magnesium ions, as well as amino acids, by facilitated diffusion, thus, it also regulates carbohydrate and lipid metabolism. Upon insulin binding to its receptor (IR), insulin receptor substrates (IRS) are phosphorylated, enhancing their binding affinity to phosphoinositide 3-kinase (PI3K). PI3K, a lipid kinase crucial for glucose uptake and metabolism, is composed of a regulatory (p85) and a catalytic subunit (p110). Once activated, PI3K phosphorylates the cell surface lipid phosphatidylinositol 4,5-bisphosphate (PIP2), generating phosphatidylinositol 3,4,5-trisphosphate (PIP3), a critical second messenger [43]. PIP3 recruits signaling proteins with pleckstrin homology (PH) domains, including phosphoinositide-dependent kinase 1 (PDK1) and AKT, activating downstream signaling pathways [43]. AKT phosphorylates multiple targets, such as FOXO, GSK3, BAD, TXNIP, and AS160, regulating processes including cell proliferation, glucose uptake, metabolism, and apoptosis [43,44]. AKT activation, for instance, inactivates FOXO via cytosolic sequestration, implicated in various cancers [44], and inactivates GSK3, regulating glycogen storage [43,44]. Moreover, AKT regulates TXNIP, influencing glucose transporter localization and glucose uptake [43]. PI3K signaling also activates the mammalian target of rapamycin (mTOR), which controls gene transcription, protein synthesis, cell proliferation [43], and tumor metabolism [45]. Additionally, downstream of PI3K, the RAS–MAPK pathway modulates critical proteins promoting survival and proliferation—such as hypoxia-inducible factor (HIF), cyclin D1, and Myc—and facilitates glucose transporter translocation, thereby further enhancing glucose uptake [43,46]. Insulin signaling also exerts AKT-independent effects, for instance, via PDK1-mediated phosphorylation of Polo-like kinase 1 (PLK1), promoting cell cycle progression [43]. Moreover, IRSs mediate protein-protein interactions, recruiting other proteins like the “growth factor receptor-bound protein 2 (Grb2)” to the binding motif on IR, to form a complex that then phosphorylates and activates RAS [41]. The phosphorylated RAS (RAS-GTP) subsequently activates the “mitogen-activated protein kinase (MAPK)” signaling cascade, including “extracellular signal-regulated kinase 1/2 (ERK1/2)” which mainly accounts for the regulation of proliferation, differentiation, cell survival, apoptosis, and stress response [47,48]. Another important IR downstream signaling pathway is Rac1, a member of the superfamily of small guanosine triphosphatases [41]. Rac 1 serves as a critical regulator of insulin-stimulated glucose uptake [49,50] and glucose-induced insulin secretion [51]. Moreover, the up-regulation of Rac1 is linked to angiogenesis, as well as tumor progression by promoting cell proliferation and migration in multiple types of cancer [52,53,54,55]. This can be justified by the structural rearrangement induced by insulin’s activation of RAC, which creates a physically conducive state for cell migration or division and enhances glycolysis and ATP production by releasing actin-bound aldolase [43]. As mentioned above, insulin can regulate cellular growth and metabolism by affecting different pathways. Activating mutations in such pathways have been linked to different diseases, including CLOVES syndrome and various types of cancer, including breast, uterine, head and neck, and colon cancers [56]. Taking this into account, it should be emphasized that insulin signaling is vital for maintaining glucose balance and normal cell function and survival. However, this same pathway must be regulated and maintained within limits, as its excessive activation can promote tumor cell proliferation and survival, and therefore can exert a significant impact on oncogenesis and chemotherapy efficacy [43]. Interestingly, insulin can even promote growth in cancer cells with low expression of IR. Such cells lacking IR are still able to make use of insulin for growth via IGF-IR. Thus, both could be targeted for developing therapeutic options [39]. Building on the previous understanding, insulin has the potential of tumor growth under certain circumstances. However, insulin also plays a pivotal role in sensitizing multiple types of tumors to chemotherapy. Insulin could be used as a biologic response modifier. Commonly known as “Insulin Potentiation Therapy (IPT)” or “Insulin Potentiation Targeted Low Dose (IPTLD)”, this technique selectively targets malignant cells while preserving normal ones, leading to significantly reduced treatment toxicity and an enhanced quality of life as reported by patients [57]. Thus, insulin is considered a double-edged sword in the battle against cancer. Results from previous research support the role of insulin in potentiating chemotherapy. Insulin pretreatment of colon cancer cells resulted in significantly enhanced susceptibility to a wide range of commonly used chemotherapeutics. It also showed an increase in the apoptosis ratio upon the co-administration of insulin with the tested drugs. Further in vivo analysis validated insulin’s ability to improve the therapeutic effect of FU without increasing toxicity [58]. This sensitizing effect of insulin was also seen in breast cancer. Combination insulin with 5FU led to a significant reduction in cell viability of the MCF-7 cell line in comparison to 5FU monotherapy. Similarly, combining Cyclophosphamide (CPA) with insulin significantly reduced cell viability compared to CPA alone. Notably, lower doses of both 5-FU and CPA inhibited MCF-7 cell growth only when cells were pretreated with insulin [59].
As discussed earlier, insulin is a critical growth factor that promotes cell growth and survival through tyrosine kinase receptor cascades and downstream pathways, such as PI3K/AKT. These pathways are frequently dysregulated in cancer, driving the invasive and rapidly growing tumor phenotype. However, when insulin’s action is combined with chemotherapeutics, such as pre-treating cancer cells with insulin, it has been shown to significantly reduce the expression of key oncogenic substrates like PIK3CA and GRB2. PIK3CA, the second most frequently mutated oncogene, is critical in cancer cell signaling, while GRB2 regulates cellular proliferation and differentiation (Figure 2). Studies demonstrate that insulin pre-treatment reduces the mRNA and protein levels of these targets, further disrupting the GRB2-MAPK pathway and initiating apoptosis [58]. This finding highlights the potential of insulin to act as a therapeutic adjunct, synergizing with chemotherapy to enhance its efficacy. 4.1. Mechanisms of Insulin Potentiation Therapy One key mechanism by which insulin sensitizes cancer cells to chemotherapeutics is through the upregulation of pro-apoptotic and autophagic proteins. For example, in breast cancer cells treated with insulin and cyclophosphamide (CPA), the expression of caspase-3, a key apoptotic marker, was significantly enhanced compared to chemotherapy alone. Additionally, insulin combined with CPA increased the levels of autophagy-related protein ATG-7, reflecting a higher proportion of cells undergoing apoptosis [59]. Similar effects were observed in colon cancer cells (SW480 and Caco-2) treated with insulin and thioglycosides, where flow cytometry analysis confirmed an increased apoptotic cell ratio compared to treatment with the drug alone [60]. 4.2. Insulin Inhibits Cell Motility Insulin also impairs cancer cell motility, a key factor in metastasis. In breast cancer cells, insulin pretreatment combined with chemotherapy reduced cell migration and proliferation, as evidenced by wound-healing assays. For instance, while CPA or 5-FU alone slightly hindered wound closure, the addition of insulin significantly amplified this effect. Similarly, in colon cancer cells treated with insulin and thioglycosides, migration and wound closure were significantly inhibited, further supporting the role of insulin in restricting metastatic potential [59,60]. 4.3. Insulin Induces GLUT-1 and GLUT-3 Expression Cancer cells rely heavily on glucose metabolism to meet their energy demands, and insulin plays a critical role in enhancing glucose uptake by increasing the expression of glucose transporters (GLUT-1 and GLUT-3). Immunocytochemistry analysis revealed that insulin significantly elevated GLUT-1 and GLUT-3 protein levels in breast cancer cells (MCF-7) and colon cancer cells (SW480 and Caco-2) [59,60]. This increase in GLUT expression, mediated via the PI3K/AKT pathway, facilitates greater glucose uptake and metabolic activity. Furthermore, the elevated GLUT expression may enhance the uptake of chemotherapeutic drugs, contributing to the observed increase in cytotoxicity. This mechanism underpins insulin’s ability to selectively sensitize cancer cells to chemotherapy, as malignant cells preferentially uptake glucose and associated drugs compared to normal cells. 4.4. Insulin Modulates Survivin Phosphorylation Another mechanism through which insulin sensitizes cancer cells to chemotherapeutics involves the modulation of survivin, a member of the inhibitor of apoptosis protein (IAP) family. Survivin’s anti-apoptotic activity depends on its phosphorylation at Thr34. In choriocarcinoma cells (JEG-3 and JAR), insulin was shown to suppress Thr34 phosphorylation of survivin in a dose-dependent manner, enhancing the apoptotic effects of 5-FU [61]. Further validation using a survivin Thr34 mutant (T34M) demonstrated that mutating this phosphorylation site reversed the insulin-induced chemotherapy sensitivity. These findings confirm that insulin’s sensitizing effect is mediated through survivin modulation, making it a promising therapeutic target [62]. Figure 3 summarizes the molecular mechanisms underlying IPT and highlights its potential to synergize with ER stress modulation, thereby enhancing the therapeutic efficacy and responsiveness of cancer cells to chemotherapy.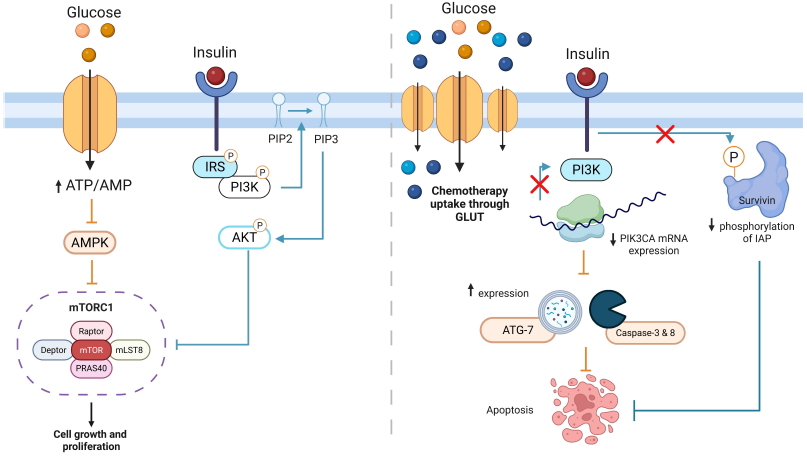
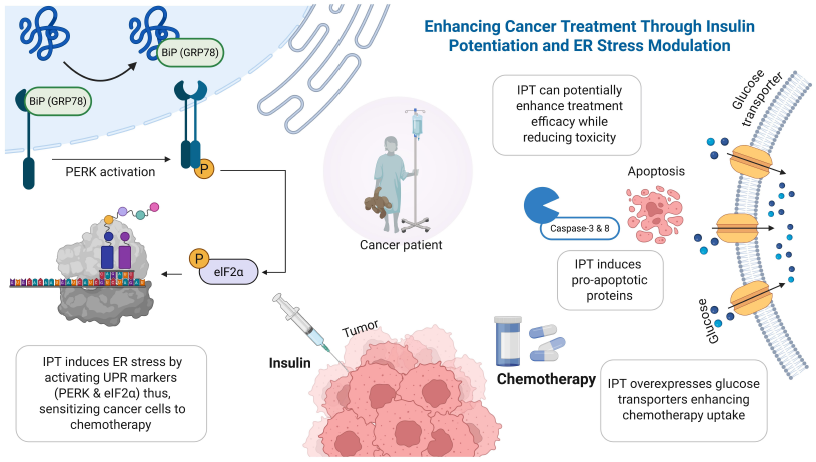
To the best of our knowledge, no research papers have directly investigated the synergistic effect between IPT and endoplasmic reticulum ER stress in cancer therapy. However, based on existing studies on the mechanisms of IPT and ER stress, it can be hypothesized that a synergistic effect may exist. IPT’s modulation of insulin could exacerbate the already elevated ER stress experienced by cancer cells due to their rapid growth and abnormal protein synthesis. Consequently, IPT may amplify ER stress within cancer cells, potentially promoting apoptosis. This heightened stress might activate the UPR, leading to cell death if the stress becomes unsustainable. Additionally, cancer cells are known to express more insulin receptors than normal cells. Exposure to insulin in IPT may facilitate greater drug absorption by cancer cells, enhancing the therapeutic effect [63]. Together, these mechanisms could synergistically increase cancer cell death. This section will emphasize more into these aspects, which are supported by different studies, as shown in Table 1. Key Studies Exploring Insulin Potentiation Therapy (IPT) and ER Stress Mechanisms in Cancer Therapy. Compared to standard chemotherapy or single-pathway treatments, combining Insulin Potentiation Therapy (IPT) with ER stress modulation offers a more targeted and potentially effective approach [67]. Chemotherapy alone often damages healthy cells and leads to drug resistance over time [68]. IPT helps improve drug uptake by using the overexpression of insulin receptors on cancer cells, allowing lower doses with fewer side effects [20]. ER stress-targeting therapies, such as PERK or IRE1 inhibitors, can induce apoptosis; however, their effectiveness may be limited by compensatory survival signals within the UPR. When used together, IPT may amplify the stress within cancer cells, making them more vulnerable to ER stress inducers. This combined strategy could drive cancer cells beyond the threshold of adaptation, leading to irreversible cell death. It also supports the move toward personalized cancer treatment by focusing on unique metabolic and proteostatic weaknesses in tumors [69]. If validated through clinical studies, this approach may improve outcomes in aggressive or resistant cancers while reducing harm to normal tissues.Table 1:
Study
Key Findings
Limitations
Mechanism of Action
Uma Kant Misra et al. [64]
Insulin induces ER stress in murine macrophages, activating UPR markers (GRP78, XBP-1, PERK, eIF2α) and enhancing antiapoptotic proteins (Bcl-2, XIAP), promoting cell survival.
Findings in murine macrophages may not fully translate to human physiology; lack direct assessment of functional consequences on apoptosis or immune responses in vivo.
Accelerated protein synthesis triggers UPR activation and antiapoptotic signaling.
Inageda et al. [65]
Insulin stabilizes ATF4 via PI3K, upregulating GRP78 to reduce ER stress-induced apoptosis. Cancer cells under IPT may experience adaptive failure, leading to apoptosis.
Primarily based on in vitro experiments, lacks in vivo validation and clinical relevance
Stabilization of ATF4 enhances GRP78 expression through PI3K signaling.
Damyanov et al. [20]
IPT combined with low-dose chemotherapy achieved 50% partial tumor regression and 25% disease stabilization in castration-resistant prostate cancer.
Small sample size and lack of a control group limit the strength of conclusions; requires further validation in larger, randomized trials.
Insulin increases drug absorption in cancer cells via elevated insulin receptor expression.
Lasalvia-Prisco et al. [66]
Combination of insulin and methotrexate significantly reduced tumor growth and improved stable disease rates in metastatic breast cancer.
Lacks a large sample size and long-term follow-up; requires further validation in randomized clinical trials.
Insulin enhances drug uptake and amplifies stress-induced apoptosis.
6.1. Insulin’s Role in Modulating ER Stress The synergistic effect of IPT may be explained by studies demonstrating insulin’s influence on ER stress. Misra et al. showed that insulin induces ER stress in murine peritoneal macrophages by accelerating global protein synthesis. This activates the UPR, with key markers such as GRP78, XBP-1, phosphorylated PERK, and eIF2α significantly upregulated, comparable to responses induced by tunicamycin, a classical ER stress inducer. Furthermore, insulin enhances antiapoptotic signaling by increasing proteins like Bcl-2, XIAP, phosphorylated CREB, and FKHR, contributing to the reduction of ER stress and the promotion of cellular survival [64]. However, in cancer cells already under heightened stress, this adaptive capacity may diminish, potentially leading to apoptosis. The additional stress from insulin could overwhelm cancer cells, enhancing the therapeutic effect by pushing them toward apoptosis. Similarly, Inageda et al. demonstrated that insulin modulates ER stress in human neuroblastoma cells by stabilizing activating ATF4 via a PI3K-dependent mechanism. This stabilization upregulates GRP78, a key chaperone that supports the UPR by promoting proper protein folding and alleviating ER stress [70]. While insulin’s role in mitigating ER stress-induced apoptosis enhances survival in normal cells, cancer cells may react differently due to their significant ER stress from rapid growth and abnormal protein synthesis. Instead of promoting survival, the additional stress from insulin may overwhelm their adaptive capacity, triggering apoptosis. 6.2. Clinical Applications and Evidence of IPT IPT may enhance the effects of traditional chemotherapeutics, especially under conditions of heightened ER stress. By increasing drug uptake and concentration within cancer cells, through upregulated insulin and insulin-like growth factor receptors, IPT could synergistically improve chemotherapeutic efficacy while minimizing dosage, thus reducing the adverse effects commonly associated with chemotherapy [20]. Ongoing clinical trials aim to further validate these observations and define the most effective IPT protocols. For instance, sixteen patients with metastatic prostate tumors were divided into two treatment groups. One group received low-dose chemotherapy comprising Epirubicin, Vinblastine, and Cyclophosphamide alongside an LHRH (Luteinizing Hormone-Releasing Hormone) agonist, while the other group was treated with low-dose Docetaxel combined with an LHRH agonist. The results showed that 50% of patients experienced a partial response, and 25% achieved disease stabilization. Importantly, no significant side effects were observed, and there were no lethal cases [20]. In another study, thirty women with metastatic breast cancer, resistant to fluorouracil, Adriamycin, cyclophosphamide, and hormone therapy, were divided into three treatment groups. One group was treated with a combination of insulin and methotrexate, another with methotrexate alone, and the third with insulin alone. The groups receiving either methotrexate alone or insulin alone predominantly showed progressive disease. In contrast, the group treated with the combination of insulin and methotrexate exhibited a higher frequency of stable disease. Moreover, the median increase in tumor size was significantly lower in the combination group compared to the groups treated with either drug alone [66]. While several studies support the role of insulin in enhancing chemotherapy efficacy, conflicting evidence exists, particularly in insulin-resistant conditions. Insulin resistance has been associated with altered signaling pathways, which may reduce insulin’s ability to potentiate anticancer effects or even promote tumor progression and metastasis in certain contexts [71]. Therefore, further investigations are needed to clarify the impact of insulin resistance on treatment outcomes and to identify patient populations that may benefit most from this approach.
Insulin Potentiation Therapy (IPT) is a promising alternative cancer treatment that combines insulin administration with low-dose chemotherapy to enhance the efficacy of anticancer drugs while minimizing side effects. Despite its innovative approach, IPT faces significant challenges that need to be addressed through rigorous research. One of the primary limitations of IPT is the limited number of studies validating its effectiveness, which hinders its clinical application. While the theoretical foundation of IPT suggests that insulin increases the permeability of cancer cells to chemotherapy drugs, this mechanism remains largely unproven [20]. Contradictory findings in existing studies further underscore the need for robust investigations to substantiate IPT’s mechanisms and therapeutic efficacy. Additionally, implementing experimental therapies such as IPT in cancer treatment requires heightened caution due to the potential risks to patient safety [72]. In addition to these challenges, combining IPT with ER stress modulation might introduce further complexities that require careful consideration. A key concern is the potential for heightened toxicity in normal cells, particularly those with high protein synthesis demands, as excessive ER stress may disrupt homeostasis and induce unintended apoptosis [73]. The dual roles of the UPR in promoting both cell survival and apoptosis could also create a risk of unpredictable therapeutic outcomes, particularly if the treatment window is not precisely defined. Insulin’s systemic effects, such as hypoglycemia and mitogenic stimulation, may be amplified when used alongside agents that enhance cellular stress [74]. Tumor heterogeneity presents another obstacle, as not all cancers exhibit elevated ER stress or overexpress insulin receptors, which may limit the generalizability of this combination approach [75]. Therefore, careful optimization of dosage, timing, and patient selection will be critical to minimize adverse effects and maximize therapeutic efficacy. Addressing these limitations may involve exploring IPT’s potential synergistic effects with pathways involved in ER stress. Targeting key markers like glucose-regulated protein 78 (GRP78) and C/EBP homologous protein (CHOP) offers a promising direction for combining IPT with strategies that exploit cancer cells’ vulnerabilities. GRP78, a molecular chaperone, is frequently overexpressed in tumors and is associated with enhanced cell survival, angiogenesis, metastasis, and poor patient outcomes [76]. Conversely, CHOP, a pro-apoptotic transcription factor activated under prolonged or severe ER stress, serves as a tipping point that triggers apoptosis in cancer cells when stress becomes unmanageable [77]. This dual role highlights the complexity of targeting ER stress in cancer therapy. While GRP78 disruption can interfere with cancer cells’ protective mechanisms, CHOP activation can selectively induce apoptosis in tumor cells. Combining IPT with metabolic modulators or agents that induce ER stress represents an exciting therapeutic avenue. Research shows that inducing ER stress can promote apoptosis in cancer cells, and this effect may be amplified when combined with metabolic interventions. For instance, targeting cancer cells’ metabolic vulnerabilities, such as glycolysis inhibitors or mitochondrial disruptors, could synergize with ER stress inducers to create a dual-targeting strategy for impairing cancer cell survival. These combination therapies could exploit cancer cells’ reliance on altered metabolic and protein-folding pathways, paving the way for innovative and potentially transformative treatment approaches [78]. To validate the synergy hypothesis, future research should include mechanistic in vitro studies using co-treatments with insulin and ER stress modulators in various cancer cell lines, followed by in vivo validation in tumor-bearing models. Additionally, well-designed clinical trials, such as randomized controlled studies comparing standard chemotherapy versus chemotherapy with insulin potentiation in patients with defined metabolic profiles, would help establish the clinical relevance of this approach.
Although IPT is an innovative approach, it faces several challenges. Notably, there is a limited body of evidence supporting its effectiveness, and the claim that insulin increases the permeability of cancer cells to chemotherapy drugs remains largely unproven [19]. Contradictory findings further complicate its clinical relevance, and the associated risks to patients have not been thoroughly investigated [67]. To address these limitations, one promising avenue is to explore the potential synergy between IPT and ER stress modulation. For instance, targeting key regulators of the unfolded protein response, such as the molecular chaperone GRP78 or the pro-apoptotic transcription factor CHOP, could enhance IPT’s therapeutic impact [20,68]. Additionally, combining glycolysis inhibitors or mitochondrial disruptors with ER stress inducers may establish a dual-targeting strategy that more effectively impairs cancer cell survival [69]. In conclusion, IPT remains insufficiently validated, with limited clinical evidence and no direct studies examining its integration with ER stress modulation. Future research should prioritize exploring these synergistic strategies to improve the efficacy of cancer therapies. These efforts hold the potential to unlock IPT’s full therapeutic value, ultimately transitioning it from an experimental concept into a validated clinical strategy for cancer therapy.
| 5-FU | 5-Fluorouracil |
| AAU | Al Ain University |
| AKT | Protein Kinase B |
| AMPK | AMP-Activated Protein Kinase |
| aHSCT | Autologous Hematopoietic Stem Cell Therapy |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| ATP | Adenosine Triphosphate |
| ATG7 | Autophagy-Related Gene 7 |
| BAD | Bcl-2-associated death promoter |
| BiP (GRP78) | Binding Immunoglobulin Protein (Glucose-Regulated Protein 78) |
| Bcl-2 | B-Cell Lymphoma 2 |
| CagA | Cytotoxin-Associated Gene A |
| CHOP | C/EBP Homologous Protein |
| CPA | Cyclophosphamide |
| CREB | cAMP Response Element-Binding Protein |
| DAPI | 4′,6-Diamidino-2-Phenylindole |
| DMSO | Dimethyl Sulfoxide |
| Dox | Doxorubicin |
| ECAR | Extracellular Acidification Rate |
| eIF2α | Eukaryotic Translation Initiation Factor 2 Alpha |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | Endoplasmic Reticulum |
| ERAD | Endoplasmic Reticulum-Associated Degradation |
| ERK1/2 | Extracellular Signal-Regulated Kinases 1 And 2 |
| FAK | Focal Adhesion Kinase |
| FDA | Food and Drug Administration |
| FOXO | Forkhead Box O |
| GADD34 | Growth Arrest and DNA Damage-Inducible Protein 34 |
| GEM | Gemcitabine |
| GLUT | Glucose Transporter |
| GPR78 | Glucose-Regulated Protein 78 |
| GRB2 | Growth Factor Receptor-Bound Protein 2 |
| GSK3 | Glycogen Synthase Kinase 3 |
| H292 | Human Lung Mucoepidermoid Carcinoma Cell Line |
| HIF-2α | Hypoxia-Inducible Factor 2 Alpha |
| HSP | Heat Shock Protein |
| IC50 | Half Maximal Inhibitory Concentration |
| ICD | Immunogenic Cell Death |
| IDH1/2 | Isocitrate Dehydrogenase 1 and 2 |
| IGF | Insulin-Like Growth Factor |
| IGF-1R | Insulin-Like Growth Factor 1 Receptor |
| IPT | Insulin Potentiation Therapy |
| IPTLD | Insulin Potentiation Targeted Low Dose |
| IRE1α | Inositol-Requiring Enzyme 1 Alpha |
| IRS | Insulin Receptor Substrate |
| JAK | Janus Kinase |
| JEG-3 | Human Placental Choriocarcinoma Cell Line |
| JNK | c-Jun N-Terminal Kinase |
| LHRH | Luteinizing Hormone-Releasing Hormone |
| MDA-MB-231 | Human Triple-Negative Breast Cancer Cell Line |
| MAPK | Mitogen-Activated Protein Kinase |
| MDSCs | Myeloid-Derived Suppressor Cells |
| MIA | Pancreatic Cancer Cell Line |
| MMPs | Matrix Metalloproteinases |
| mTOR | Mammalian Target of Rapamycin |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide Assay |
| MYC | MYC Proto-Oncogene |
| OCR | Oxygen Consumption Rate |
| OGT | O-GlcNAc Transferase |
| PANC | Pancreatic Cancer Cell Line |
| PDI | Protein Disulfide Isomerase |
| PDK1 | Phosphoinositide-Dependent Kinase 1 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PDCD4 | Programmed Cell Death Protein 4 |
| PERK | Protein Kinase R-Like ER Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| PIP2 | Phosphatidylinositol 4,5-Bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-Trisphosphate |
| PLK1 | Polo-Like Kinase 1 |
| PRKAA1 | AMP-Activated Protein Kinase Catalytic Subunit Alpha-1 |
| PTEN | Phosphatase and Tensin Homolog |
| qPCR | Quantitative Polymerase Chain Reaction |
| RAC1 | Ras-Related C3 Botulinum Toxin Substrate 1 |
| RIDD | Regulated IRE1-Dependent Decay |
| RNA-seq | RNA Sequencing |
| ROS | Reactive Oxygen Species |
| RUNX1 | Runt-Related Transcription Factor 1 |
| S6K | Ribosomal Protein S6 Kinase |
| SDH | Succinate Dehydrogenase |
| SH2 | Src Homology 2 Domain |
| SOD2 | Superoxide Dismutase 2 |
| T34M | Survivin Thr34-Mutant |
| TCA | Tricarboxylic Acid Cycle |
| TXNIP | Thioredoxin-Interacting Protein |
| UPR | Unfolded Protein Response |
| XBP1 | X-Box Binding Protein 1 |
| XIAP | X-Linked Inhibitor Of Apoptosis Protein |
N.A.-D. and M.S. conducted the literature review, contributed to the original manuscript writing, and created the figures. S.K. conducted the literature review, drafted the original manuscript. W.K.M., M.K. and G.K. provided critical insights, assisted in refining the manuscript, and ensured scientific rigor. R.G. conceptualized the review, supervised the project, guided the manuscript structure and content, and performed the final revisions. All authors read and approved the final version of the manuscript.
The authors declare no conflicts of interest regarding this manuscript.
Not applicable.
Not applicable.
[1] Navarro, C.; Ortega, Á.; Santeliz, R.; Garrido, B.; Chacín, M.; Galban, N.; Vera, I.; De Sanctis, J.B.; Bermúdez, V. Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches. Pharmaceutics 2022, 14. [CrossRef]
[2] Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [CrossRef]
[3] Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism as a Therapeutic Strategy for Cancer. Exp. Mol. Med. 2023, 55, 706–715. [CrossRef] [PubMed]
[4] Tufail, M.; Jiang, C.H.; Li, N. Altered Metabolism in Cancer: Insights into Energy Pathways and Therapeutic Targets. Mol. Cancer 2024, 23, 203. [CrossRef] [PubMed]
[5] Kumar, R.; Mishra, A.; Gautam, P.; Feroz, Z.; Vijayaraghavalu, S.; Likos, E.; Shukla, G.C.; Kumar, M. Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy. Cancers 2022, 14. [CrossRef]
[6] Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [CrossRef] [PubMed]
[7] Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21. [CrossRef]
[8] Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [CrossRef]
[9] Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic Reticulum Stress Signals in the Tumour and Its Microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [CrossRef]
[10] Yu, M.; Lun, J.; Zhang, H.; Wang, L.; Zhang, G.; Zhang, H.; Fang, J. Targeting UPR Branches, a Potential Strategy for Enhancing Efficacy of Cancer Chemotherapy. Acta Biochim. Biophys. Sin. 2021, 53, 1417–1427. [CrossRef]
[11] Beilankouhi, E.A.V.; Sajadi, M.A.; Alipourfard, I.; Hassani, P.; Valilo, M.; Safaralizadeh, R. Role of the ER-Induced UPR Pathway, Apoptosis, and Autophagy in Colorectal Cancer. Pathol. Res. Pract. 2023, 248, 154706. [CrossRef] [PubMed]
[12] Zhang, W.; Shi, Y.; Oyang, L.; Cui, S.; Li, S.; Li, J.; Liu, L.; Li, Y.; Peng, M.; Tan, S.; et al. Endoplasmic Reticulum Stress—A Key Guardian in Cancer. Cell Death Discov. 2024, 10, 343. [CrossRef]
[13] Ron, D.; Hubbard, S.R. How IRE1 Reacts to ER Stress. Cell 2008, 132, 24–26. [CrossRef] [PubMed]
[14] Glimcher, L.H.; Lee, A.H.; Iwakoshi, N.N. XBP-1 and the Unfolded Protein Response (UPR). Nat. Immunol. 2020, 21, 963–965. [CrossRef]
[15] Almeida, L.M.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. The PERKs of Mitochondria Protection during Stress: Insights for PERK Modulation in Neurodegenerative and Metabolic Diseases. Biol. Rev. 2022, 97, 1737–1748. [CrossRef]
[16] Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [CrossRef] [PubMed]
[17] Talukdar, G.; Orr, H.T.; Lei, Z. The PERK Pathway: Beneficial or Detrimental for Neurodegenerative Diseases and Tumor Growth and Cancer. Hum. Mol. Genet. 2023, 32, 2545–2557. [CrossRef]
[18] Lei, Y.; Yu, H.; Ding, S.; Liu, H.; Liu, C.; Fu, R. Molecular Mechanism of ATF6 in Unfolded Protein Response and Its Role in Disease. Heliyon 2024, 10, e25937. [CrossRef]
[19] Lei, Z.N.; Tian, Q.; Teng, Q.X.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z. Understanding and Targeting Resistance Mechanisms in Cancer. MedComm 2023, 4, e265. [CrossRef]
[20] Damyanov, C.; Gerasimova, D.; Maslev, I.; Gavrilov, V. Low-Dose Chemotherapy with Insulin (Insulin Potentiation Therapy) in Combination with Hormone Therapy for Treatment of Castration-Resistant Prostate Cancer. ISRN Urol. 2012, 2012, 1–6. [CrossRef]
[21] Zarrabi, A.; Perrin, D.; Kavoosi, M.; Sommer, M.; Sezen, S.; Mehrbod, P.; Bhushan, B.; Machaj, F.; Rosik, J.; Kawalec, P.; et al. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers 2023, 15. [CrossRef] [PubMed]
[22] Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [CrossRef] [PubMed]
[23] Salvagno, C.; Mandula, J.K.; Rodriguez, P.C.; Cubillos-Ruiz, J.R. Decoding Endoplasmic Reticulum Stress Signals in Cancer Cells and Antitumor Immunity. Trends Cancer 2022, 8, 930–943. [CrossRef] [PubMed]
[24] Liao, H.; Liu, S.; Ma, Q.; Huang, H.; Goel, A.; Torabian, P.; Mohan, C.D.; Duan, C. Endoplasmic Reticulum Stress Induced Autophagy in Cancer and Its Potential Interactions with Apoptosis and Ferroptosis. Biochim. Biophys. Acta Mol. Cell Res. 2025, 1872. [CrossRef]
[25] Unal, B.; Kuzu, O.F.; Jin, Y.; Osorio, D.; Kildal, W.; Pradhan, M.; Kung, S.H.Y.; Oo, H.Z.; Daugaard, M.; Vendelbo, M.; et al. Targeting IRE1α Reprograms the Tumor Microenvironment and Enhances Anti-Tumor Immunity in Prostate Cancer. Nat. Commun. 2024, 15, 8895. [CrossRef]
[26] Samanta, S.; Yang, S.; Debnath, B.; Xue, D.; Kuang, Y.; Ramkumar, K.; Lee, A.S.; Ljungman, M.; Neamati, N. The Hydroxyquinoline Analogue YUM70 Inhibits GRP78 to Induce ER Stress–Mediated Apoptosis in Pancreatic Cancer. Cancer Res. 2021, 81, 1883–1895. [CrossRef]
[27] Wu, Y.; Xie, Q.; Wu, L.; Li, Z.; Li, X.; Zhang, L.; Zhang, B. Identification of Activating Transcription Factor 6 (ATF6) as a Novel Prognostic Biomarker and Potential Target in Oral Squamous Cell Carcinoma. Gene 2024, 915, 148436. [CrossRef]
[28] Mandula, J.K.; Chang, S.; Mohamed, E.; Jimenez, R.; Sierra-Mondragon, R.A.; Chang, D.C.; Obermayer, A.N.; Moran-Segura, C.M.; Das, S.; Vazquez-Martinez, J.A.; et al. Ablation of the Endoplasmic Reticulum Stress Kinase PERK Induces Paraptosis and Type I Interferon to Promote Anti-Tumor T Cell Responses. Cancer Cell 2022, 40, 1145–1160.e9. [CrossRef]
[29] Shao, A.; Xu, Q.; Kang, C.W.; Cain, C.F.; Lee, A.C.; Tang, C.H.A.; Del Valle, J.R.; Hu, C.-C.A. IRE-1-Targeting Caged Prodrug with Endoplasmic Reticulum Stress-Inducing and XBP-1S-Inhibiting Activities for Cancer Therapy. Mol. Pharm. 2022, 19, 1059–1067. [CrossRef]
[30] Xu, W.; Shen, Y. Curcumin Affects Apoptosis of Colorectal Cancer Cells through ATF6-Mediated Endoplasmic Reticulum Stress. Chem. Biol. Drug Des. 2024, 103. [CrossRef]
[31] Li, X.; Zheng, J.; Chen, S.; Meng, F.-D.; Ning, J.; Sun, S.-L. Oleandrin, a Cardiac Glycoside, Induces Immunogenic Cell Death via the PERK/elF2α/ATF4/CHOP Pathway in Breast Cancer. Cell Death Dis. 2021, 12, 314. [CrossRef] [PubMed]
[32] González-Ortiz, A.; Pulido-Capiz, A.; Castañeda-Sánchez, C.Y.; Ibarra-López, E.; Galindo-Hernández, O.; Calderón-Fernández, M.A.; López-Cossio, L.Y.; Díaz-Molina, R.; Chimal-Vega, B.; Serafín-Higuera, N.; et al. eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model. Cells 2022, 11. [CrossRef] [PubMed]
[33] She, C.; Wu, C.; Guo, W.; Xie, Y.; Li, S.; Liu, W.; Xu, C.; Li, H.; Cao, P.; Yang, Y.; et al. Combination of RUNX1 Inhibitor and Gemcitabine Mitigates Chemo-Resistance in Pancreatic Ductal Adenocarcinoma by Modulating BiP/PERK/eIF2α-Axis-Mediated Endoplasmic Reticulum Stress. J. Exp. Clin. Cancer Res. 2023, 42, 238. [CrossRef] [PubMed]
[34] Yang, Z.; Huo, Y.; Zhou, S.; Guo, J.; Ma, X.; Li, T.; Fan, C.; Wang, L. Cancer Cell-Intrinsic XBP1 Drives Immunosuppressive Reprogramming of Intratumoral Myeloid Cells by Promoting Cholesterol Production. Cell Metab. 2022, 34, 2018–2035.e8. [CrossRef]
[35] Yan, Y.; He, M.; Zhao, L.; Wu, H.; Zhao, Y.; Han, L.; Wei, B.; Ye, D.; Lv, X.; Wang, Y.; et al. A Novel HIF-2α Targeted Inhibitor Suppresses Hypoxia-Induced Breast Cancer Stemness via SOD2-mtROS-PDI/GPR78-UPRER Axis. Cell Death Differ. 2022, 29, 1769–1789. [CrossRef]
[36] Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
[37] Liao, M.; Yao, D.; Wu, L.; Luo, C.; Wang, Z.; Zhang, J.; Liu, B. Targeting the Warburg Effect: A Revisited Perspective from Molecular Mechanisms to Traditional and Innovative Therapeutic Strategies in Cancer. Acta Pharm. Sin. B 2024, 14, 953–1008. [CrossRef]
[38] . In Diabetes and Breast Cancer: An Analysis of Signaling Pathways , Bala, A., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2024; . . [CrossRef]
[39] Monteiro, M.; Zhang, X.; Yee, D. Insulin Promotes Growth in Breast Cancer Cells through the Type I IGF Receptor in Insulin Receptor Deficient Cells. Exp. Cell Res. 2024, 434, 113862. [CrossRef]
[40] Giovannucci, E. Insulin, Insulin-Like Growth Factors and Colon Cancer: A Review of the Evidence. J. Nutr. 2001, 131, 3109S–3120S. [CrossRef]
[41] Leitner, B.P.; Siebel, S.; Akingbesote, N.D.; Zhang, X.; Perry, R.J. Insulin and Cancer: A Tangled Web. Biochem. J. 2022, 479, 583–607. [CrossRef]
[42] Argirion, I.; Weinstein, S.J.; Männistö, S.; Albanes, D.; Mondul, A.M. Serum Insulin, Glucose, Indices of Insulin Resistance, and Risk of Lung Cancer. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 1519–1524. [CrossRef] [PubMed]
[43] Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin–PI3K Signalling: An Evolutionarily Insulated Metabolic Driver of Cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [CrossRef]
[44] Tsai, P.J.; Lai, Y.H.; Manne, R.K.; Tsai, Y.S.; Sarbassov, D.; Lin, H.K. Akt: A Key Transducer in Cancer. J. Biomed. Sci. 2022, 29. [CrossRef]
[45] Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR Signaling Pathway and mTOR Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10. [CrossRef] [PubMed]
[46] Molinaro, A.; Becattini, B.; Mazzoli, A.; Bleve, A.; Radici, L.; Maxvall, I.; Sopasakis, V.R.; Molinaro, A.; Bäckhed, F.; Solinas, G. Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kα and PI3Kβ Activities and Is Promoted by RAS. Cell Metab. 2019, 29, 1400–1409.e5. [CrossRef] [PubMed]
[47] Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [CrossRef]
[48] Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10. [CrossRef]
[49] Audzeyenka, I.; Rogacka, D.; Rachubik, P.; Typiak, M.; Rychłowski, M.; Angielski, S.; Piwkowska, A. The PKGIα–Rac1 Pathway Is a Novel Regulator of Insulin-Dependent Glucose Uptake in Cultured Rat Podocytes. J. Cell. Physiol. 2021, 236, 4655–4668. [CrossRef]
[50] Takenaka, N.; Nakao, M.; Hasegawa, K.; Chan, M.P.; Satoh, T. The Guanine Nucleotide Exchange Factor FLJ00068 Activates Rac1 in Adipocyte Insulin Signaling. FEBS Lett. 2020, 594, 4370–4380. [CrossRef]
[51] Thamilselvan, V.; Kowluru, A. Paradoxical Regulation of Glucose-Induced Rac1 Activation and Insulin Secretion by RhoGDIβ in Pancreatic β-Cells. Small GTPases 2021, 12, 114–121. [CrossRef]
[52] Seiz, J.R.; Klinke, J.; Scharlibbe, L.; Lohfink, D.; Heipel, M.; Ungefroren, H.; Giehl, K.; Menke, A. Different Signaling and Functionality of Rac1 and Rac1b in the Progression of Lung Adenocarcinoma. Biol. Chem. 2020, 401, 517–531. [CrossRef] [PubMed]
[53] Wahoski, C.C.; Singh, B. The Roles of RAC1 and RAC1B in Colorectal Cancer and Their Potential Contribution to Cetuximab Resistance. Cancers 2024, 16. [CrossRef] [PubMed]
[54] Liang, J.; Liu, Q.; Xia, L.; Lin, J.; Oyang, L.; Tan, S.; Peng, Q.; Jiang, X.; Xu, X.; Wu, N.; et al. Rac1 Promotes the Reprogramming of Glucose Metabolism and the Growth of Colon Cancer Cells through Upregulating SOX9. Cancer Sci. 2023, 114, 822–836. [CrossRef]
[55] Yamaguchi, M.; Takagi, K.; Sato, A.; Miki, Y.; Miyashita, M.; Sasano, H.; Suzuki, T. Rac1 Activation in Human Breast Carcinoma as a Prognostic Factor Associated with Therapeutic Resistance. Breast Cancer 2020, 27, 919–928. [CrossRef]
[56] Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [CrossRef]
[57] Damyanov, C.; Gerasimova, D.; Avramova, L.; Masley, I. K Insulin Potentiation Therapy in the Treatment of Malignant Neoplastic Diseases: A Three-Year Study. J. Cancer Sci. Ther. 2012, 4, 74–78. [CrossRef]
[58] Agrawal, S.; Woźniak, M.; Łuc, M.; Makuch, S.; Pielka, E.; Agrawal, A.K.; Wietrzyk, J.; Banach, J.; Gamian, A.; Pizon, M.; et al. Insulin Enhancement of the Antitumor Activity of Chemotherapeutic Agents in Colorectal Cancer Is Linked with Downregulating PIK3CA and GRB2. Sci. Rep. 2019, 9. [CrossRef]
[59] Agrawal, S.; Łuc, M.; Ziółkowski, P.; Agrawal, A.K.; Pielka, E.; Walaszek, K.; Zduniak, K.; Woźniak, M. Insulin-Induced Enhancement of MCF-7 Breast Cancer Cell Response to 5-Fluorouracil and Cyclophosphamide. Tumor Biol. 2017, 39. [CrossRef]
[60] Agrawal, S.; Wozniak, M.; Luc, M.; Walaszek, K.; Pielka, E.; Szeja, W.; Pastuch-Gawolek, G.; Gamian, A.; Ziolkowski, P. Insulin and Novel Thioglycosides Exert Suppressive Effect on Human Breast and Colon Carcinoma Cells. Oncotarget 2017, 8, 114173–114182. [CrossRef]
[61] Martínez-García, D.; Manero-Rupérez, N.; Quesada, R.; Korrodi-Gregório, L.; Soto-Cerrato, V. Therapeutic Strategies Involving Survivin Inhibition in Cancer. Med. Res. Rev. 2019, 39, 887–909. [CrossRef]
[62] Shan, Y.; Li, Y.; Han, H.; Jiang, C.; Zhang, H.; Hu, J.; Sun, H.; Zhu, J. Insulin Reverses Choriocarcinoma 5-Fluorouracil Resistance. Bioengineered 2021, 12, 2087–2094. [CrossRef]
[63] Malaguarnera, R.; Belfiore, A. The Insulin Receptor: A New Target for Cancer Therapy. Front. Endocrinol. 2011, 2. [CrossRef]
[64] Misra, U.K.; Pizzo, S.V. Up-Regulation of GRP78 and Antiapoptotic Signaling in Murine Peritoneal Macrophages Exposed to Insulin. J. Leukoc. Biol. 2005, 78, 187–194. [CrossRef] [PubMed]
[65] Ha, D.P.; Lee, A.S. Insulin-like Growth Factor 1-Receptor Signaling Stimulates GRP78 Expression through the PI3K/AKT/mTOR/ATF4 Axis. Cell. Signal. 2020, 75, 109736. [CrossRef]
[66] Lasalvia-Prisco, E.; Cucchi, S.; Vázquez, J.; Lasalvia-Galante, E.; Golomar, W.; Gordon, W. Insulin-Induced Enhancement of Antitumoral Response to Methotrexate in Breast Cancer Patients. Cancer Chemother. Pharmacol. 2004, 53, 220–224. [CrossRef]
[67] Djamgoz, M.B.A. Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms. Cancers 2022, 14. [CrossRef]
[68] Arafat, M.; Sakkal, M.; Beiram, R.; AbuRuz, S. Nanomedicines: Emerging Platforms in Smart Chemotherapy Treatment—A Recent Review. Pharmaceuticals 2024, 17. [CrossRef] [PubMed]
[69] Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [CrossRef] [PubMed]
[70] Inageda, K. Insulin Modulates Induction of Glucose-Regulated Protein 78 during Endoplasmic Reticulum Stress via Augmentation of ATF4 Expression in Human Neuroblastoma Cells. FEBS Lett. 2010, 584, 3649–3654. [CrossRef]
[71] Ferguson, R.D.; Novosyadlyy, R.; Fierz, Y.; Alikhani, N.; Sun, H.; Yakar, S.; LeRoith, D. Hyperinsulinemia Enhances c-Myc-Mediated Mammary Tumor Development and Advances Metastatic Progression to the Lung in a Mouse Model of Type 2 Diabetes. Breast Cancer Res. 2012, 14, R8. [CrossRef]
[72] Zheng, J.Y.; Mixon, A.C.; McLarney, M.D. Safety, Precautions, and Modalities in Cancer Rehabilitation: An Updated Review. Curr. Phys. Med. Rehabil. Rep. 2021, 9, 142–153. [CrossRef] [PubMed]
[73] Yuan, S.; She, D.; Jiang, S.; Deng, N.; Peng, J.; Ma, L. Endoplasmic Reticulum Stress and Therapeutic Strategies in Metabolic, Neurodegenerative Diseases and Cancer. Mol. Med. 2024, 30, 40. [CrossRef] [PubMed]
[74] He, J.; Zhou, Y.; Sun, L. Emerging Mechanisms of the Unfolded Protein Response in Therapeutic Resistance: From Chemotherapy to Immunotherapy. Cell Commun. Signal. 2024, 22, 89. [CrossRef] [PubMed]
[75] Zhu, L.; Jiang, M.; Wang, H.; Sun, H.; Zhu, J.; Zhao, W.; Fang, Q.; Yu, J.; Chen, P.; Wu, S.; et al. A Narrative Review of Tumor Heterogeneity and Challenges to Tumor Drug Therapy. Ann. Transl. Med. 2021, 9, 1351. [CrossRef]
[76] Zheng, Y.Z.; Cao, Z.G.; Hu, X.; Shao, Z.M. The Endoplasmic Reticulum Stress Markers GRP78 and CHOP Predict Disease-Free Survival and Responsiveness to Chemotherapy in Breast Cancer. Breast Cancer Res. Treat. 2014, 145, 349–358. [CrossRef]
[77] Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 10. [CrossRef]
[78] Wolpaw, A.J.; Dang, C.V. Exploiting Metabolic Vulnerabilities of Cancer with Precision and Accuracy. Trends Cell Biol. 2018, 28, 201–212. [CrossRef]