
 ACCESS
Research Article
ACCESS
Research Article
Volume 1, Article ID: 2025.0007

Dhruva Bhat
db110499@gmail.com


Appala Venkata Ramana Murthy
avrmurthy@diat.ac.in
1 Department of Applied Physics, Defence Institute of Advanced Technology, Pune-411025, India
2 Corresponding Author: A. V. R. Murthy, Department of Applied Physics, Defence Institute of Advanced Technology, Pune, India,
* Author to whom correspondence should be addressed
Received: 13 Dec 2024 Accepted: 10 Apr 2025 Available Online: 11 Apr 2025 Published: 30 Jun 2025
Lipid ordering plays a crucial role in maintaining the mechanical strength of the lipid bilayer, which is influential in controlling biological functions. This is associated with the well-known phenomena of “phase state behavior” of the bilayer. The significant factors that influence phase state behavior are stimuli such as cholesterol, physiological conditions like temperature and pH, and the structure of the lipid molecule itself, which can differ either in the head group or the acyl chain. The cholesterol concentration influences the phase state behavior and the ordering of the lipid bilayer. In this paper, we investigate the behavior of Phosphatidylcholine lipids with cholesterol, using Coarse-Grained Molecular Dynamics Simulation. The simulation is performed using a CGMartini force field. The production run of all successful simulations is completed with the same biological condition with a total runtime of 12 µs. We chose four lipids namely DLPC, POPC, DGPC, and DIPC with varied acyl chain chemical structures and investigated the influence of cholesterol. We have calculated bilayer thickness, area per lipid, order parameter, radial distribution function, and partial density which are a measure of bilayer phase state behavior. The results indicate efficient cholesterol packing in DLPC, while POPC exhibits variations due to its asymmetric acyl chain structure, which contains a single double bond in one of the chains. In contrast, DGPC and DIPC display lower order parameters, attributed to the presence of multiple double bonds in their acyl chains, which disrupt ordered packing. This study elucidates the importance of the acyl chain structure and the cholesterol composition on the phase state behavior of the lipid membrane.
The expansion of Molecular Dynamics (MD) simulation [1,2] has become a necessary tool in the study of biomolecules, helping to understand the complex membrane physics, such as molecular interactions and dynamics [3]. To design an effective drug delivery system, it is important to understand the dynamic behavior of lipid membranes where MD simulation comes into play [4]. MD simulation techniques are proven to be effective in the study of neurodegenerative and cardiovascular diseases, by investigating the role of membrane disordering and disruption [5,6,7]. They are also effective in investigating the biophysical and chemical properties of lipids and their membranes, such as protein and molecule interactions, and the effect on the lipid structure and permeability by factors like temperature, pressure, and pH [8,9]. The advance of MD simulation led to the development of many buildings and analyzing tools such as GROMACS [10], VMD [11], FATSlim [12], and GridMAT-MD [13]. Recent advancements in the study of biological cell membranes revealed the presence of many lipids and their bilayers which are essential for cellular molecular activities. Among them, one of the major and most abundant phospholipids in the biological membranes is Phosphatidylcholine (PC). As the name suggests, it has a simple structure where a glycerol backbone connects between the two fatty acid chains and a phosphocholine headgroup [14]. PC lipids play a major role in assisting cellular membrane activities, also maintaining membrane fluidity and stability by providing a protective and supportive layer to the biological cell and membrane protein [15,16]. The different structural properties of fatty acid chains have a major significance on their membrane. Unsaturated and saturated fatty acids influence membrane fluidity differently: the former increases fluidity, while the latter decreases it, resulting in a more rigid membrane. [17]. PC lipids play a key role in cellular dynamics such as cell signaling pathways [18]. They also can be hydrolyzed by phospholipases to generate secondary messengers [19]. In the process of lipid metabolism and homeostasis, PC lipids act as precursors [20]. Due to the wide range of fatty acid chains, these lipids exhibit well-defined structural and physical properties, which significantly influence membrane interactions [21]. Examples include 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) with two 16-carbon chains and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) with two 12-carbon chains. This unique property of PC lipids made them one of the most used model membranes for MD simulations study and also to design liposomes and other drug delivery systems [22,23]. As mentioned before, modifications in PC lipid composition and its metabolism are directly related to cardiovascular, liver, and neurodegenerative diseases [24,25,26]. Understanding the properties of these lipids is crucial for developing the desired medications. The artificial phospholipids with varying acyl chain structure [27], 1,2-dilauroyl-rac-glycero-3-phosphocholine (DLPC), 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), 1,2-di-(11Z-eicosenoyl)-sn-glycero-3-phosphocholine (DGPC), and 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DIPC) are ideal models for the study of lipid bilayers and membrane physics in MD simulations. Analyzing these simulations helps in understanding the biophysical characteristics and dynamic behavior of biological membranes which are crucial in mapping the disease probe membranes [28,29]. The influence of sterols like cholesterol on bilayers is essential for preserving the fluidity, structure, and functionality of membranes. They are abundant in mammalian cell membranes and majorly contribute to controlling membrane dynamics and linked proteins [30,31]. By studying cholesterol-filled lipid bilayers through MD simulations, researchers can gain a deeper understanding of cholesterol’s influence on the membrane. This study is vital in the field of medicine to develop medication and therapies for cholesterol-related diseases [32,33]. Kamble et al. studied the condensation effect caused by the cholesterol with lipid bilayer. They confirmed the result with the help of Spectroscopic Ellipsometry (SE) and MD simulation. Their successful report verified the interaction of cholesterol on the Lipid bilayer. Another published result with the Atomic Force Microscope (AFM) supported the results on asymmetricity, phase separation, and phase state behavior of lipid bilayer [34,35]. The Cholesterol affecting the high Tm Lipid monolayers upsetting the phase state was studied by Murthy et al. [36]. The internal properties such as temperature show a major contribution to the behavior of phase and change in the order parameter with cholesterol in polar lipids [37]. Chiu et al. studied the unusual behavior of lipid cross-sectional area with the increase in sterol concentration [38]. The effects of the order parameter and tilt angle of cholesterol and lipid hydrocarbon chains, along with hydrogen bond interactions, were studied and confirmed by Robinson et al. [39]. Many published results confirm the MD simulation study of the order parameter of phospholipid’s acyl chain [40,41,42]. A popular coarse-grained (CG) model for effectively simulating biomolecular systems while maintaining crucial molecular features is the MARTINI force field [43]. This helps in grouping atoms into larger particles, making complex compounds simpler, which results in reducing the computing time for the successful simulation. In the GROMACS [10], the MARTINI force field [43] works very well for modeling lipid bilayers and sterols. The advancement in computational biology significantly contributes to the study of complicated biological structures and their interactions with different lipids and proteins, and also to understanding membrane dynamics. By mimicking certain biological conditions through simulation, it is possible to create and duplicate the exact biological activity which will be similar to the experiment. The results obtained through these complex simulations can be compared with the experimental results for a better understanding of the complexity of the membrane. In this paper, we have investigated various lipid bilayers with varying the chemical structure of the acyl chain and the cholesterol interaction with these bilayers with varying concentrations. The study specifically targets structural properties like bilayer thickness, area per lipid, order parameters, and dynamic properties like radial distribution function and partial density. The results obtained not only show dependence on the percentage of cholesterol but also the chemical structure of the acyl chain. We have also compared the results obtained across the four lipid bilayers—DLPC, POPC, DGPC, and DIPC—not only among themselves but also with findings reported in the literature, as summarized in the table below (Table 1). These results can be used to predict experimental properties like asymmetry-induced topographical and nano-mechanical properties [34,35,44,45]. Comparison of reported studies with the current simulation results.
Table 1:
Authors
Year
Work Done Related to Biological Membranes
Simulation Methodology
Major Details of Lipids Investigated
(Thickness (nm),
APL (nm2))
Chiu et al. [38]
2002
Cholesterols effect on biological phospholipid membrane.
All-atom MD simulation
DPPC + Chol (50%)
(APL: ~0.45 nm2)
Pantelopulos et al. [46]
2018
Phase change behavior in lipid membrane with cholesterol.
Coarse-Grained MD simulation
__
Ermilova et al. [47]
2019
Behavior of cholesterol inside the phospholipid membrane.
All-atom MD simulation
PC + Chol
(14:0–14:0 + 50%)
(APL: 0.40 nm2)
Shahane et al. [48]
2019
Investigation of physical properties of the artificial lipid bilayer.
All-atom MD simulation
PC + Chol (50%)
(Thickness: 4.45 nm,
APL: 0.45 nm2)
Khakbaz et al. [49]
2018
Study of phase transition behavior in PC lipids.
All-atom MD simulation
Pure DMPC
(APL: 0.52 ± 0.04 nm2)
Pure DPPC
(APL: 0.49 ± 0.02 nm2)
Chakraborty et al. [50]
2020
Cholesterols affect the physical properties of the unsaturated lipid bilayer.
All-atom MD simulation
DOPC + Chol (50%)
(Thickness: 3.99 nm,
APL: ∼0.44 nm2)
Dutta et al. [51]
2025
The trans-bilayer motion of cholesterol in the lipid bilayer.
All-atom and Coarse-Grained MD simulation
POPS system
(Thickness: 4.14 ± 0.06 nm,
APL: 0.56 ± 0.011 nm2)
Plasma membrane
(Thickness: 4.18 ± 0.04 nm,
APL: 0.51 ± 0.06 nm2)
Zhang et al. [52]
2015
Cholesterols effect on phase behavior in DPPC.
Coarse-Grained MD simulation
DPPC + Chol (50%)
(APL: ~0.43 nm2)
Kučerka et al. [53]
2008
Change in bilayer thickness in the bilayer due to cholesterol’s impact.
All-atom MD simulation
diC22:1PC + Chol (40%)
(Thickness: 2.4 nm,
APL: 0.81 nm2 (varying))
Alwarawrah et al. [54]
2010
Impact of cholesterol on the PC lipid bilayer and study on condensation effect.
All-atom MD simulation
DOPC + Chol (50%)
(Thickness: 4.5 nm,
APL: 0.40 ± 0.01 nm2)
Kamble et al. [35]
2020
Effect of interleaflet decoupling due to induced cholesterol
Coarse-Grained MD simulation
DOPC + Chol (50%)
(Thickness: 5.16 nm,
APL: 0.59 nm2)
This work
-
The effect of cholesterol and acyl chain of lipid on the structural property of bilayer
Coarse-Grained MD simulation
DLPC + Chol (50%)
(Thickness: 4.01 nm,
APL: 0.39 nm2)
POPC + Chol (50%)
(Thickness: 4.12 nm,
APL: 0.42 nm2)
DGPC + Chol (50%)
(Thickness: 4.6 nm,
APL: 0.42 nm2)
DIPC + Chol (50%)
(Thickness: 3.73 nm,
APL: 0.47 nm2)
2.1. MD Simulation Details All the simulations are completed using GROMACS 2018.1 and 2024.1 versions. The necessary GROMACS file (.gro) and topology (.top) files are generated with the Insane (INSert membrANE) [55] tool, which uses the forcefield parameter from the Martini force field. A simulation box with dimensions 15 nm x 10 nm x 9 nm, containing lipids and sterol molecules was generated. The concentration of Na⁺ and Cl⁻ ions in the simulation was maintained at 0.15 M to mimic physiological ionic conditions and ensure charge neutrality, thereby providing a stable environment for the lipid bilayer under investigation. The output configuration is written to .gro file in a cubic periodic boundary condition, solvated with water. The same conditions were maintained throughout the experiment. The portable topology files (.itp) (Martini V2.0) which contain the information on all the necessary parameters required for the simulation were taken from the cgmartini website. The topology (.top) file was written using the result generated by the Insane tool and required .itp files were included. In each leaflet of the bilayer, an exact number of lipids is maintained (Table 2). Before the start of the simulation, energy minimization and equilibration steps were performed. Energy minimization was performed using the steepest descent algorithm, with a maximum of 1000 steps, using the Verlet cutoff scheme for non-bonded interactions and applying periodic boundary conditions in all dimensions. Coulombic interactions were controlled with the reaction-field method, truncating interactions at 1.1 nm and setting the relative dielectric constant to 15. Van der Waals interactions used a cutoff and potential-shift, with a cutoff distance of 1.1 nm. For equilibration, the Martini force field was used to stabilize the system at the desired temperature and pressure. Time steps of 1 fs, 5 fs, 10 fs, and 30 fs were employed. A Berendsen thermostat maintained the temperature at 298 K with a 1 ps time constant, and pressure was set to 1 bar with a 6 ps time constant. Initial velocities were generated at 298 K, and constraints were applied using the LINCS algorithm. Confirming effective minimization and equilibration run, a newly configured .mdp file was used to initiate the production run. The integrator algorithm chosen is the standard MD integrator with a time step of 0.03 ps and a total of 400 million steps, corresponding to an overall simulation time of 12 µs. Centre of mass motion removal is configured to occur every 100 steps. For energy minimization, a convergence tolerance of 10 and an initial step size of 0.01 are specified, with a maximum of 20 iterations allowed. The steepest descent method is used with corrections applied every 1000 steps. Neighbor searching employs the Verlet cutoff scheme, updating the neighbor list every 20 steps, with a cutoff radius of 1.0 nm. Electrostatics are managed using the reaction-field method with a Coulomb cutoff of 1.1 nm and a relative dielectric constant of 15. Van der Waals interactions are treated with a cutoff of 1.1 nm, without long-range dispersion corrections. Temperature coupling is managed via the v-rescale method with a reference temperature of 300 K and a time constant of 1 ps. Pressure coupling is semi-isotropic using the Parrinello-Rahman method with a reference pressure of 1 bar and a compressibility of 3 × 10−4/bar, with a time constant of 12 ps. Velocities are generated at the start of the simulation corresponding to a temperature of 300 K. The LINCS algorithm is used for constraint solving with an order of 8 and 2 iterations. No user-defined groups, walls, or external electric fields were applied, ensuring a straightforward and unbiased molecular dynamics simulation setup with the use of MPI (Message Passing Interface), the simulation is performed on multiple processors simultaneously, enhancing performance and reducing simulation time. To ensure consistent conditions in every simulation, the same mdp files are used for energy minimization, equilibration, and production runs. Simulation parameters for varying cholesterol concentrations in lipid bilayers. 2.2. Simulation Analysis An energy file (.edr), a GROMACS compressed trajectory file (.xtc), and a lossless trajectory file (.trr) were generated as a post-simulation output. The .xtc file contains compressed atomic coordinates at various time steps, making it useful for trajectory visualization and analysis. In contrast, the .trr file stores full-precision, lossless trajectory data, including atomic coordinates, velocities, and forces, and is primarily used for more detailed analyses. This level of data is required for performing precise calculations and detailed analysis. The .edr file records the simulation’s configurations, including integrator settings and time steps, providing a reference for the simulation parameters. Together, these files offer complete post-simulation analysis, enabling tasks like visualizing molecular dynamics to perform accurate computational calculations. Area per lipid (APL) provides valuable insights into the structural properties, and stability of lipid membranes, also understanding sterols interaction, the packing density of the bilayer, and its behavior under different conditions, which are important for understanding cellular developments and designing membrane-targeted drugs. A direct method of APL calculation can be done using the inbuilt GROMACS tool gmx_energy. By determining the area of the simulation box and dividing it by the total number of lipids in the single leaflets assuming both have an equal number of lipids. FATSLiM v0.2.1 tool uses a different approach to calculate APL by completely identifying lipids and membranes initially. Each lipid has an available area, and the area per lipid may be roughly calculated using the average of the accessible areas. As a result, it calculates the membrane area which is the average of the two leaflet areas, and the leaflet area is just the total of all individual areas [12]. GridMAT-MD 2.0 (Grid-based Membrane Analysis Tool for use with Molecular Dynamics) tool, reads from a coordinate file (.gro) which only has a single frame [13]. A parameter file is fed into the system in which the user has to define the input parameter file, reference point, solvent, ions, number of grid points across each axis, and the type of grid to be generated as an output i.e., column, matrix, or vector. The output is analyzed to get the result of APL. The Bilayer Thickness (BT) study of a lipid membrane is essential because it influences membrane permeability and the function of embedded sterols. Understanding bilayer thickness helps elucidate the physical properties of the membrane and its role in various biological processes, such as signaling and transport. For example, a thicker bilayer may stabilize certain receptor conformations, enhancing or inhibiting their ability to transmit signals, also may make it harder for small molecules to diffuse. GROMACS tool gmx density is used for the calculation of BT. Provide the proper index file by choosing the phosphate beads as the headgroup. The generated output can be analyzed using XMGrace software (Version: Grace-5.1.25), where the bilayer thickness (BT) is determined by measuring the distance between the headgroup peak positions. Rather than change in the input index file and reference atom, both FATSLiM and GridMAT-MD use similar approaches for the thickness calculation [12,13]. Later results from direct calculation, FATSLiM, and GridMAT-MD are compared. Studying Partial density is important because it provides insights into the spatial distribution of different components within a system. This information is crucial for understanding the structural and dynamic properties of complex mixtures, such as lipid bilayers or sterol solutions. By analyzing partial densities, one can determine how molecules are arranged and how they interact with each other. Here also the GROMACS gmx density tool is used in the calculation. Investigating the Order parameter of lipid and sterol simulations is crucial for understanding the degree of lipid tail alignment and membrane fluidity. Defined as To generate the output, a Python script do-order-gmx5.py is used within the GROMACS environment. The script is called by giving. xtc and .tpr files as input along with the start and end time for analysis (in ps), specific frequency (in steps) at which the order parameters are calculated, type of lipid, and number of lipids in the leaflet. The calculation is performed along the z-axis and averaged over a specified number of simulation steps. The resulting output, including the profile data, can be used to compare with published results. Radial Distribution Functions (RDF) is an important method for understanding how molecules are arranged around a reference point in a system. It provides a detailed analysis of molecular packing, local structure, and how different molecules interact with each other. For example, in lipid bilayers or sterol mixtures, RDF analysis helps in understanding how lipids interact with each other or with cholesterol. In this study, we used the inbuilt GROMACS tool gmx rdf to analyze these interactions and better understand the structural and organizational characteristics of the system. Later the generated results were visualized using XMGrace software(Version: Grace-5.1.25). A representative simulation result of the lipid bilayer with various types of lipids i.e., DLPC, POPC, DGPC, and DIPC, with 50% cholesterol, was shown in Figure 1a–d, and bead representation of PC lipid and cholesterol was shown in Figure 1e. This will provide a visual overview of the cholesterol distribution in the lipid bilayer.
Table 2:
Sr No.
Lipid
Cholesterol Concentration
No Lipids in One Bilayer
No Cholesterols in One Bilayer
Water Molecules
Ions (Na++Cl−)
01.
DLPC
50
123
123
6373
140
40
148
98
6372
140
30
172
74
6374
140
20
197
49
6374
140
10
222
24
6375
140
00
247
0
6371
140
02.
POPC
50
123
123
5793
128
40
148
98
5676
124
30
172
74
5568
122
20
197
49
5527
122
10
222
24
5360
118
00
247
0
5366
118
03.
DGPC
50
123
123
5529
122
40
148
98
5406
120
30
172
74
5328
118
20
197
49
5213
114
10
222
24
5154
114
00
247
0
5096
112
04.
DIPC
50
123
123
5822
128
40
148
98
5677
124
30
172
74
5635
124
20
197
49
5458
120
10
222
24
5471
120
00
247
0
5243
116
where in MARTINI, θ represents the angle between the coarse-grained tail bead vector and the bilayer normal, rather than atomic C-H bonds. This approach adapts the concept of order parameters to a coarse-grained level of detail, while still offering insights into lipid alignment and bilayer organization.
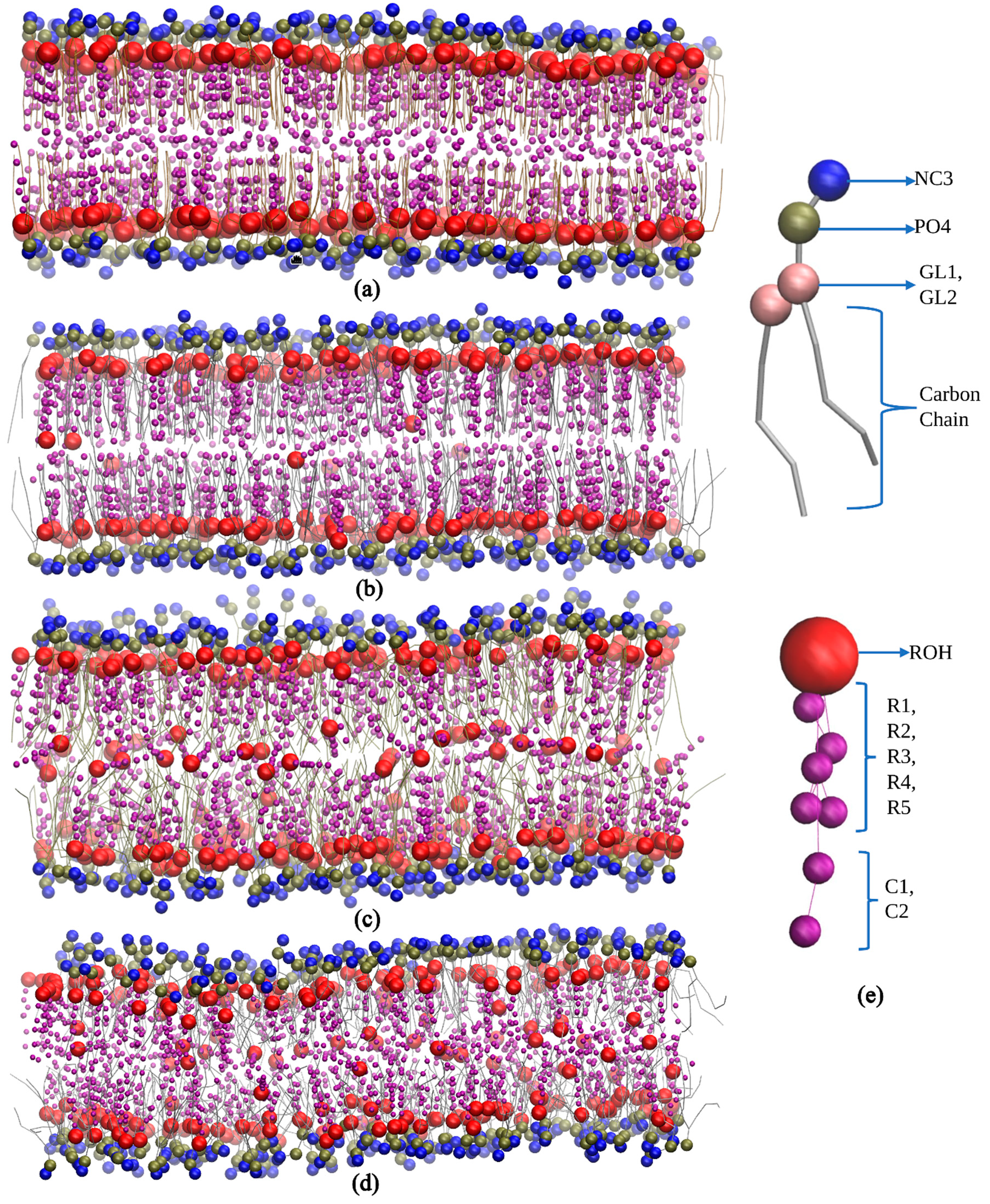
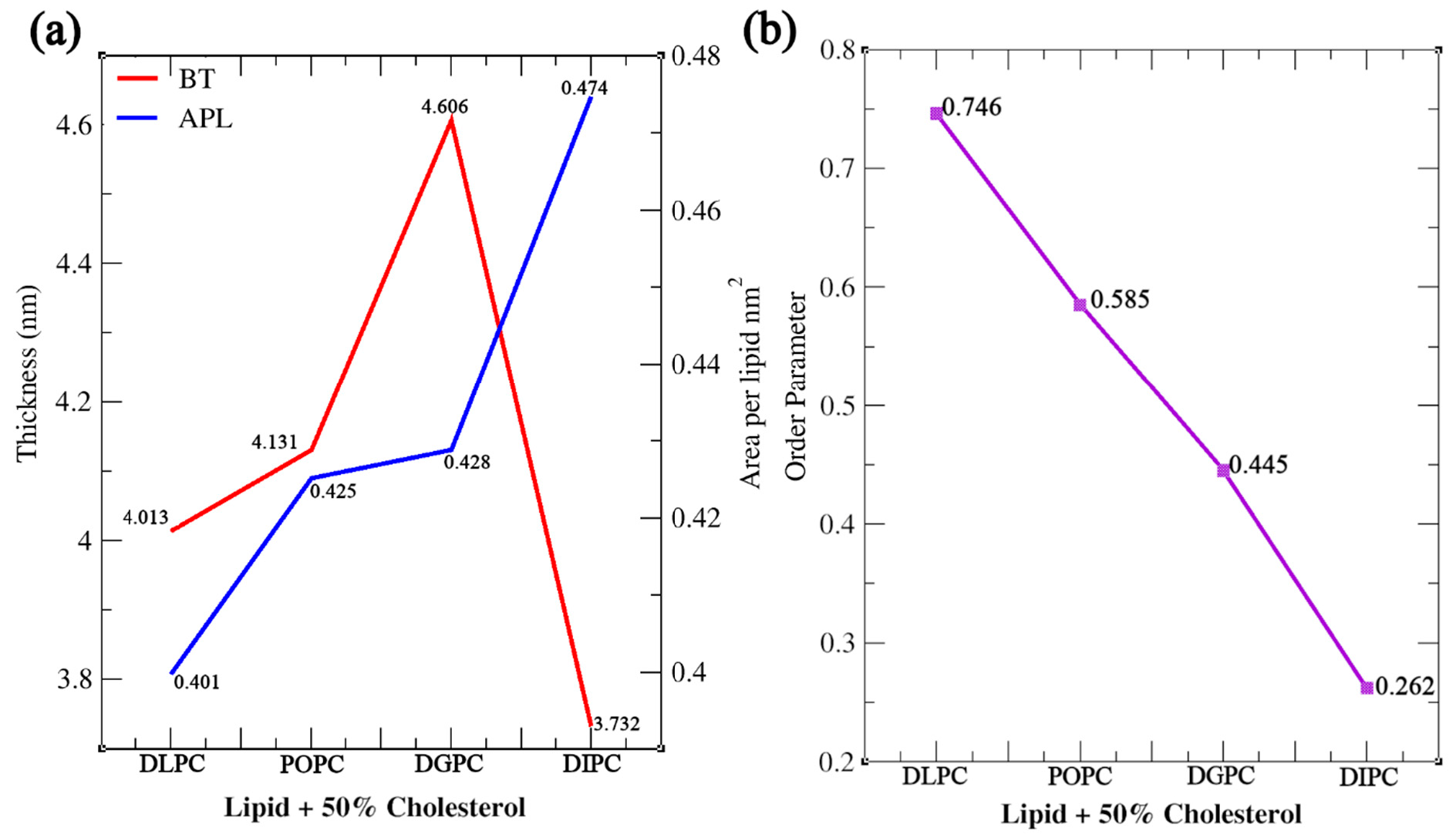
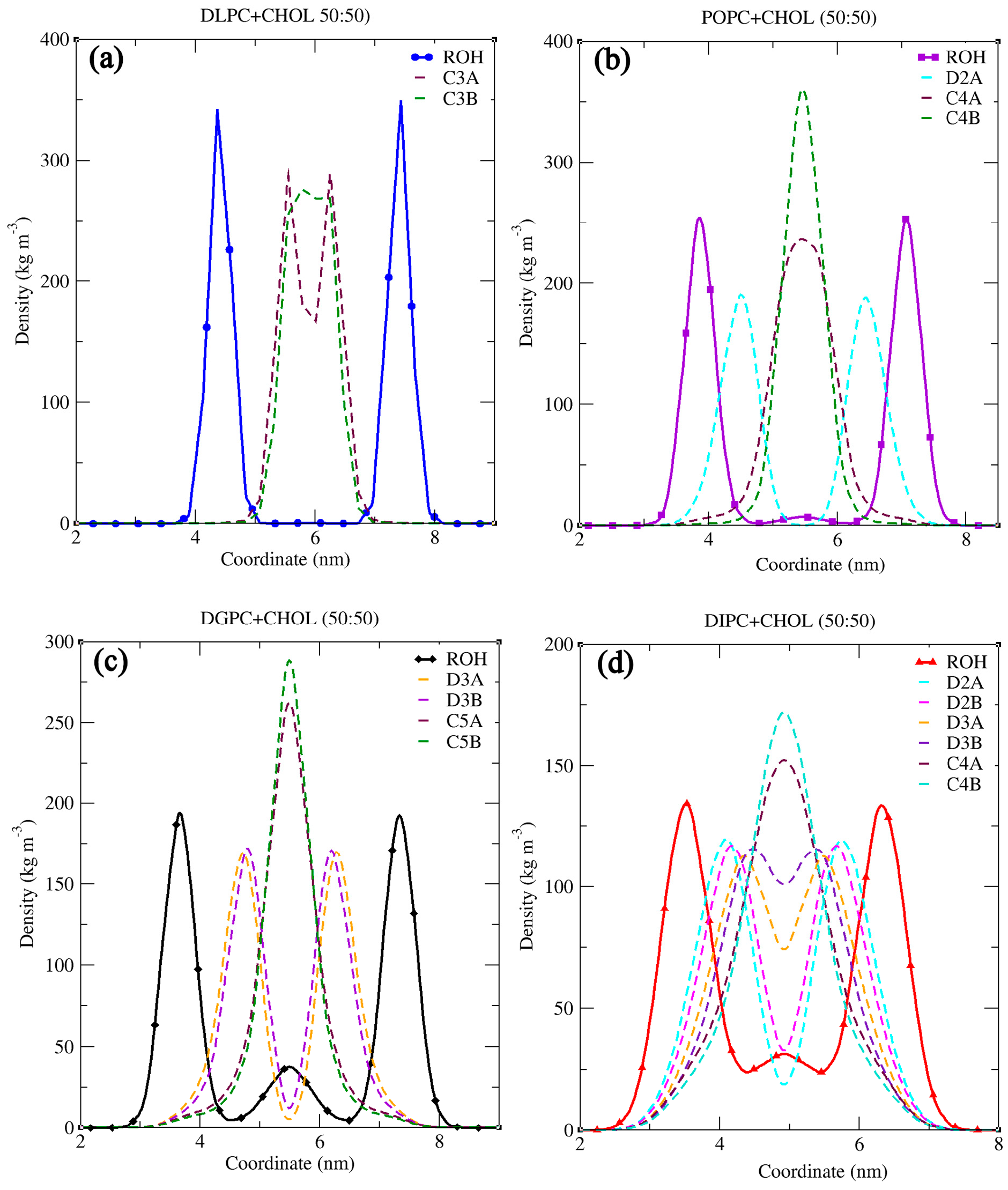
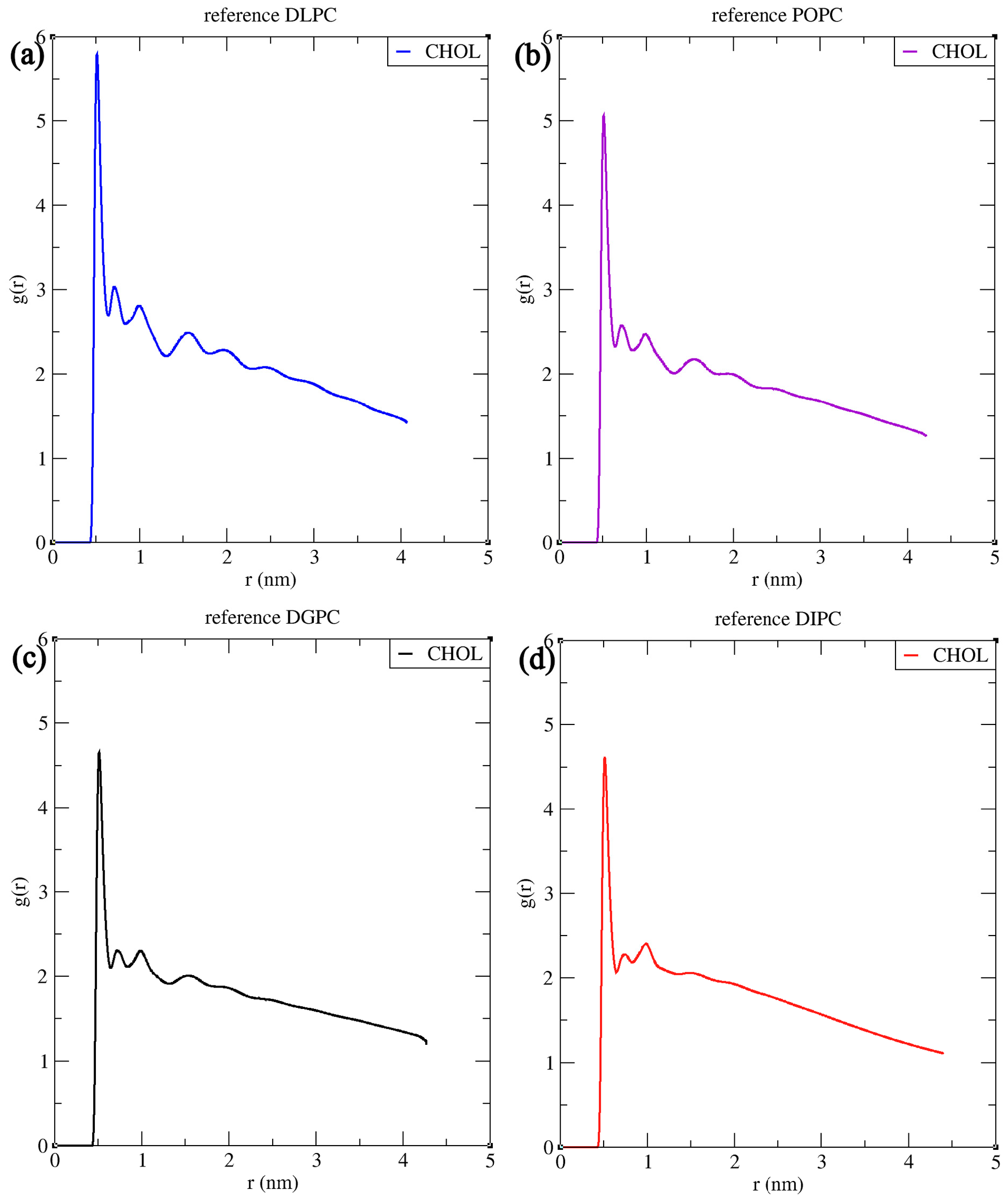
3.1. Structural Properties of Phosphatidylcholine Lipids and Cholesterol The structural configuration of phosphatidylcholine lipids: 1,2-dilauroyl-rac-glycero-3-phosphocholine (DLPC), 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), 1,2-di-(11Z-eicosenoyl)-sn-glycero-3-phosphocholine (DGPC), and 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DIPC) were analyzed. Since these molecules (DLPC, POPC, DGPC, and DIPC) are all phospholipids, they share a central structure of glycerol, a phosphate group linked to choline, and two fatty acid chains attached to the glycerol. DLPC has two short, saturated lauroyl chains (12 carbons each) (Figure 2a), POPC has one saturated palmitoyl chain (16 carbons) and one unsaturated oleoyl chain (18 carbons with a double bond) (Figure 2b). DGPC has two longer, unsaturated eicosenoyl chains (20 carbons with a double bond at the 11th position) (Figure 2c), and DIPC features two linoleoyl chains (18 carbons with two double bonds each) (Figure 2d). In the .itp files, the atoms are represented in terms of beads. Below is the detailed comparative explanation of molecules as written in the respected .itp files. The positively charged nitrogen atom (NC3) represented by the atom type Q0, bonded to a phosphate group (PO4) with the atom type Qa. The two neutral glycerol atoms (GL1 and GL2) are represented by the atom type Na. In DLPC each fatty acid chain is represented by three beads: C1A, C2A, and C3A for one chain, and C1B, C2B, and C3B for the other, all of type C1, indicating that they are simple, neutral, and do not have any branching or double bonds. POPC has more complex chains, the first chain starts with a carbon atom of type C1 (C1A), followed by a carbon of type C3 (D2A) (which indicates a double bond), and then continues with two more carbon atoms of type C1 (C3A and C4A). The second hydrocarbon chain is simpler, consisting of four consecutive carbon atoms of type C1 (C1B, C2B, C3B, C4B). In DGPC, the backbone connects to two hydrocarbon chains, each chain starts with a carbon atom (C1A and C1B) of type C1, continues with a sequence of carbon atoms (C2A, D3A, C4A, C5A for the first chain, and C2B, D3B, C4B, C5B for the second chain), where the D3A and D3B atoms are of type C3. Among the four lipids, DIPC has the most complex structure. Its first hydrocarbon chain starts with a carbon atom of type C1 (C1A), followed by two carbons of type C4 (D2A and D3A), indicating the presence of double bonds, and ends with a carbon of type C1 (C4A). The second chain has a similar structure (C1B, D2B, D3B, C4B). This structural configuration, with two extended hydrophobic tails and a hydrophilic headgroup, is characteristic of the typical arrangement of a phosphatidylcholine lipid, which is important for forming bilayer structures in membrane simulations. In cholesterol molecules, the ROH group is represented by the SP1 bead. The subsequent groups are represented by SC1 beads (R1, R3, R4, R5) and SC3 (R2) (Figure 2e). The aliphatic tail segments in cholesterol are represented by SC1 (C1) and C1 (C2) beads. Bond interactions are defined within the molecule to maintain its structure. For example, the bond between C1 and C2 has a length of 0.425 nm and a force constant of 1250. Structural integrity is further confirmed by constraints between key beads (SP1, SC1/SC3, SC1/C1). The correct spatial arrangement and stability of non-linear components in the molecule are defined with virtual sites. Dihedral angles and exclusions focus on maintaining proper orientation and stability. These configurations ensure accurate modeling of the molecular dynamics and interactions of cholesterol within lipid membranes. 3.2. Simulation Result Analysis of Different Concentrations of Cholesterol with Lipids The. xtc,. tpr,. edr, and .gro files were used to analyze the results such as bilayer thickness, area per lipid, and order parameter. Sterols are a rigid, planar ring structure that inserts between lipid molecules, promoting the extension and alignment of lipid chains, which results in lipid tails being more stretched. The data demonstrate that sterols significantly influence the structure of lipid membranes, though the extent of their effects varies across different lipid types. Sterol addition generally results in thicker bilayers, reduced lipid area, and better alignment of lipid tails, enhancing membrane stability (Table S1). However, the degree of these changes depends on the specific lipid composition, with some membranes responding more dramatically to sterol incorporation than others. The influence of sterol concentration on lipid membranes was explored across four different types: DLPC, POPC, DGPC, and DIPC. The effect of sterol concentration on bilayer thickness showed distinct trends across the different lipids. For DLPC, the thickness increased from 3.445 nm at 0% sterol to 4.013 nm at 50% (Figure 3a), reflecting the membrane’s expansion due to sterol incorporation. In POPC, the change in bilayer thickness with increasing cholesterol concentration is relatively minimal. The lowest thickness observed is 3.897 nm at 30% cholesterol, while the highest is 4.281 nm at 10%. Unlike DLPC, POPC does not exhibit a consistent increasing trend. This may be because of the presence of an unsaturated oleoyl chain in POPC (Figure 3b). DGPC and DIPC showed a more pronounced change, with bilayer thickness increasing from 4.370 nm to 4.606 nm (Figure 3c) and 3.461 nm to 3.732 nm (Figure 3d), peaking at 40% sterol concentration (4.667 nm and 3.795 correspondingly), suggesting that the effect of sterols on bilayer expansion may level off around 40-50%. The cholesterol aligns itself between the acyl chain tails more tightly due to its rigid structure, which results in a stiffer bilayer and reduces kinks, leading to a thicker bilayer [50,57]. The deformity observed at 50% cholesterol in lipids other than DLPC is attributed to phase behavior and the presence of a double bond in the lipid. The lipid tails can only be extended and aligned to a certain degree, beyond which additional sterols may not contribute to further structural changes. Sterol addition led to a decrease in the area per lipid across all lipid types, indicating tighter molecular packing within the membranes. For DLPC, the area per lipid dropped from 0.591 nm2 to 0.399 nm2 as sterol concentration increased (Figure 4a). POPC also saw a substantial reduction, from 0.642 nm2 to 0.425 nm2 (Figure 4b). DGPC’s area per lipid decreased from 0.666 nm2 to 0.428 nm2 (Figure 4c), while DIPC showed a greater decline from 0.741 nm2 to 0.474 nm2 (Figure 4d), reflecting less pronounced membrane condensation. This decrease in area per lipid significantly alters the membrane surface properties. The increase in cholesterol helps increase the stability of the membrane, for which the membrane shows resistance to changes in temperature and mechanical stress, which in turn contributes to the membrane’s structural integrity and functional efficiency. The order parameter, which reflects the alignment of lipid acyl chains, increased with rising sterol concentration in all cases; however, the extent of this increase varied among different lipid types. In DLPC, the order parameter rose significantly from 0.479 to 0.746, indicating enhanced tail alignment (Figure 5a). POPC exhibited a smaller increase, from 0.375 to 0.585 (Figure 5b). For DGPC, the order parameter climbed from 0.331 to 0.445 (Figure 5c), showing improved alignment, while DIPC experienced a more modest rise from 0.227 to 0.262 (Figure 5d), but the highest being at 40% concentration i.e., 0.272, suggesting a limited impact of sterols on tail alignment compared to the other lipids. The results are further confirmed by comparing the values obtained from different computational tools such as FATSLiM v0.2.1 and GridMAT-MD 2.0. (Table S1). 3.3. Partial Density and Radial Distribution Function (RDF) Analysis of Phosphatidylcholine Lipids Bilayer The simulation analysis showed a significant observation at the 50% cholesterol and Lipid composition. The increase in cholesterol leads to an increase in order parameters which results in the reaching the saturation point. However, in DLPC the bilayer thickness follows the increasing trend, and the order parameter is higher compared to other lipids. This proves that DLPC may still have some space in between bilayer lipid tails, till it reaches its saturation point. Figure 6a depicts the partial density of the headgroup of lipid PO4, indicating their well-defined position in the bilayer, close to the water-lipid interface. From the previously observed phenomenon of DGPC and sterol interaction i.e., a decrease in the bilayer thickness at the 50% sterol concentration, the same pattern is observed in the case of DIPC (3.732 nm) (Figure 7a), which is very low compared to other lipids. The ROH groups show broader and less defined peaks, indicating a more dispersed distribution within the bilayer (Figure 6b). The graph depicts the position of cholesterol presence in the bilayer wherein 0 represents the center of the bilayer and −2 and 2 are the two leaflets. The more stretched the ROH is towards the bilayer, the more it shows the order parameter (Figure 7b). In Figure 6b, the distribution of cholesterol within the bilayer shows an increasing order which can also be visualized in Figure 1 and can be compared with the graphs, resulting in the decrease of order parameter from DLPC to DIPC (0.746 to 0.246) (Figure 5). The cholesterol moving in-between the bilayer rather than aligning itself with the lipid headgroup is due to the presence of double bonds in the lipid tail (Figure 1). ‘DLPC allows more cholesterol to align toward the headgroup than DIPC. The double bond in the hydrophobic tail creates a kink, which increases the area per lipid but disrupts the packing of neighboring lipids. This disruption affects the bilayer’s structure, potentially hindering cholesterol’s ability to pass through. The impact of the double bond on bilayer movement can be visualized in the density map of ROH along with the double bond and terminal carbon atom of the lipid tail (Figure 7). POPC has only one double bond in a single chain (D2A) (Figure 7b), it has a lesser distribution of cholesterol towards the headgroup compared to DLPC (Figure 7b). DIPC has the highest number of double bonds (D2A, D3A, D2B, and D3B) (Figure 7d) making it have a more flexible tail, which results in the highest distribution of cholesterol between the bilayer than the other four simulated lipids. The flexibility of the acyl chain strongly influences cholesterol alignment and distribution within the lipid bilayer. Lipids like DLPC, with fully saturated chains, tend to exhibit more stretched acyl chains compared to lipids such as DIPC, which contain double bonds. An increasing number of double bonds in lipid tails reduces chain stretch and increases flexibility, particularly in the presence of cholesterol. This increased flexibility disrupts the alignment of cholesterol between the lipid’s hydrophilic headgroups, leading to lower bilayer order parameters and faster saturation of order parameter values at lower cholesterol concentrations. Figure 8 visualizes this phenomenon confirming the movement and distribution of molecules inside the bilayer depending on its structural properties. The Radial Distribution Function (RDF) graphs (Figure 9) provide a clear picture of how cholesterol interacts with different lipids—DLPC, POPC, DGPC, and DIPC—revealing the role of lipid structure in shaping the bilayer’s molecular organization. DLPC shows the strongest interaction with cholesterol (Figure 9a), as seen in the sharp, high RDF peak around 0.5 nm and consistent values beyond 1 nm. This is due to its fully saturated lipid tails, which promote tighter packing and better cholesterol alignment. On the other hand, DIPC exhibits the weakest interaction (Figure 9d), with a smaller peak and a rapid drop-off, reflecting the flexibility of its tails with multiple double bonds. These double bonds create kinks that disrupt packing and make it harder for cholesterol to align, resulting in reduced bilayer order. POPC and DGPC fall in between (Figure 9b,c), with POPC performing slightly better due to having only one double bond, causing less disruption. These findings align with observations of bilayer thickness and order, where DLPC forms a more structured bilayer, while DIPC’s flexible tails lead to a looser, more disorganized arrangement. Compared to cholesterol-lipid RDFs, lipid-lipid interactions are generally stronger and more consistent, especially for DLPC and POPC. DIPC remains the most disordered lipid in both cases, highlighting how double bonds in the lipid tails disrupt packing and reduce order. Saturated lipids like DLPC promote tight packing, while unsaturated lipids like DIPC introduce more flexibility and disorder (Figure S1). Overall, this analysis highlights how lipid tail saturation and flexibility directly affect cholesterol distribution, lipid packing, and bilayer stability.![Figure 2: <p>The chemical structure of phosphatidylcholine lipids (<strong>a</strong>) DLPC, (<strong>b</strong>) POPC, (<strong>c</strong>) DGPC, (<strong>d</strong>) DIPC, and sterols (<strong>e</strong>) cholesterol [<a href="#ref56">56</a>].</p>](/uploads/source/articles/molecular-modeling-connect/2025/volume1/20250007/image002.png)
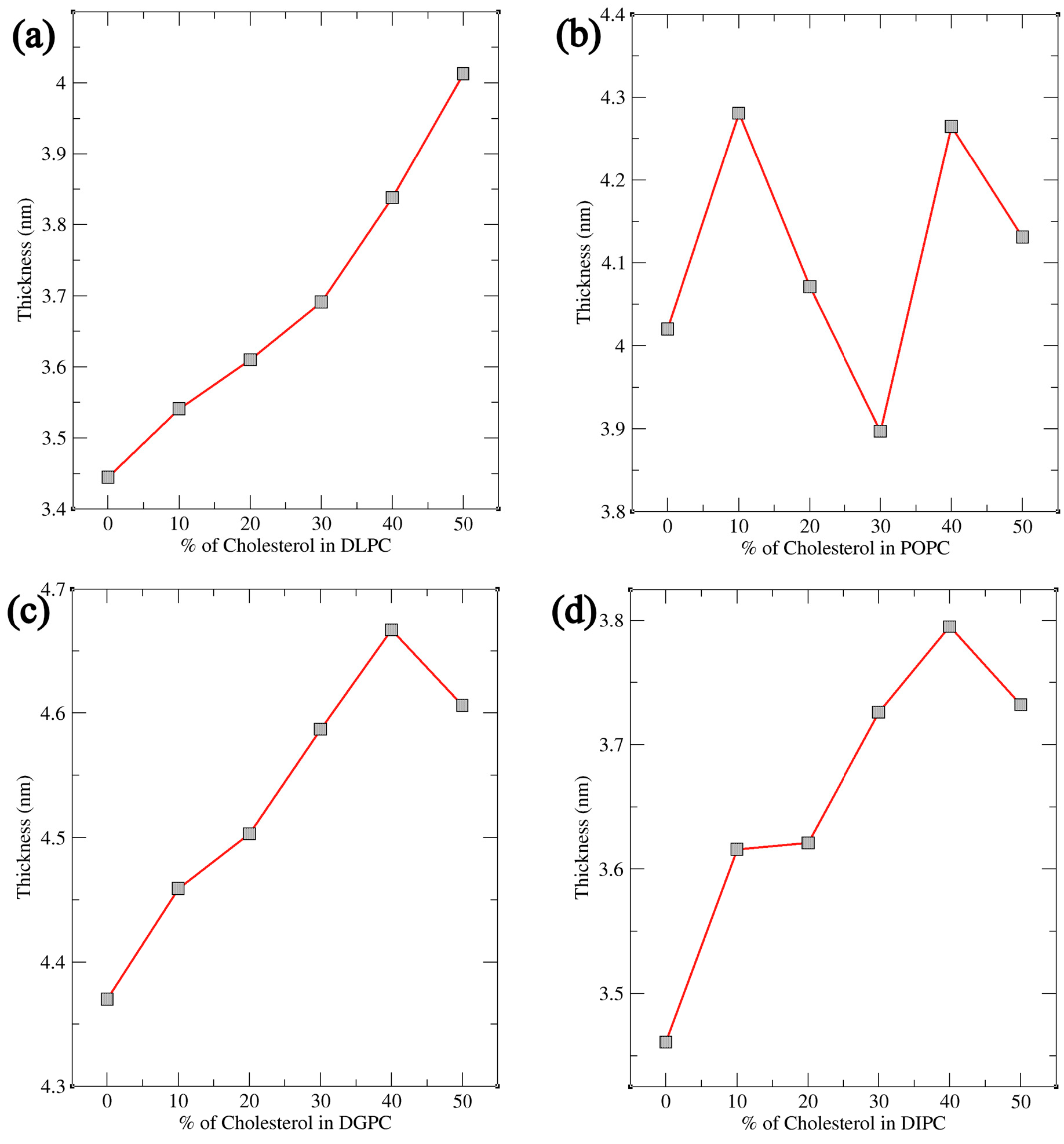
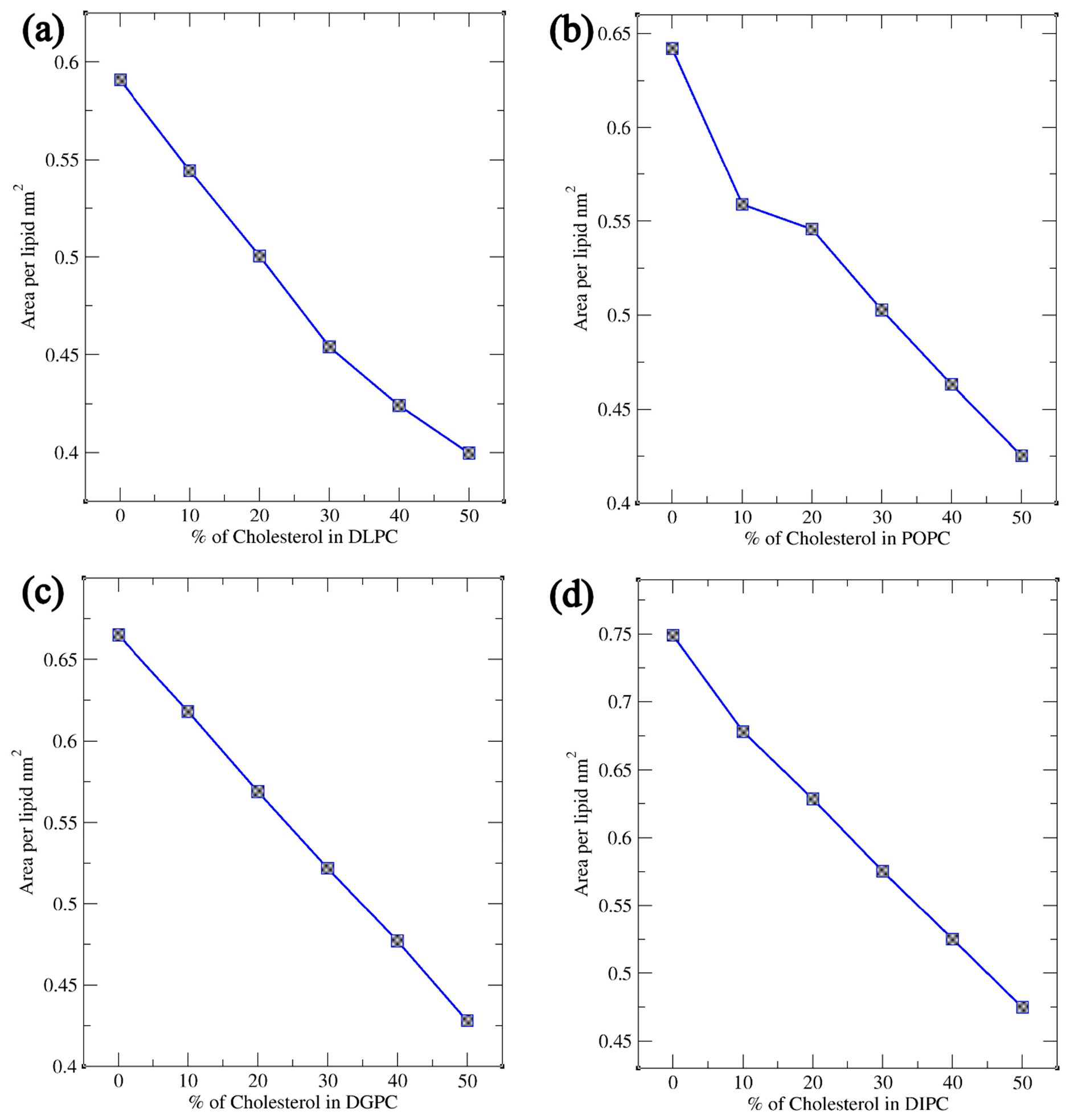
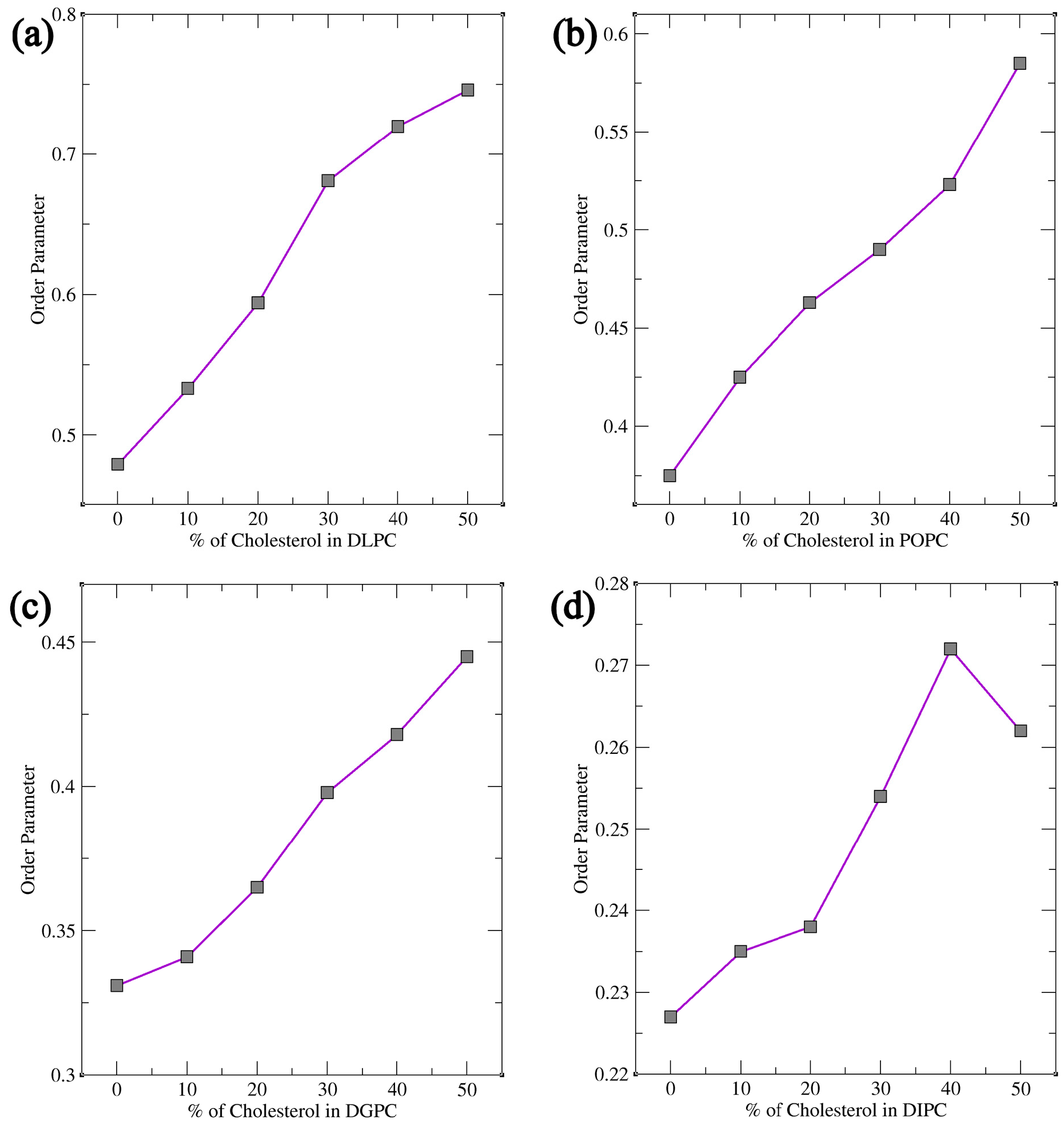
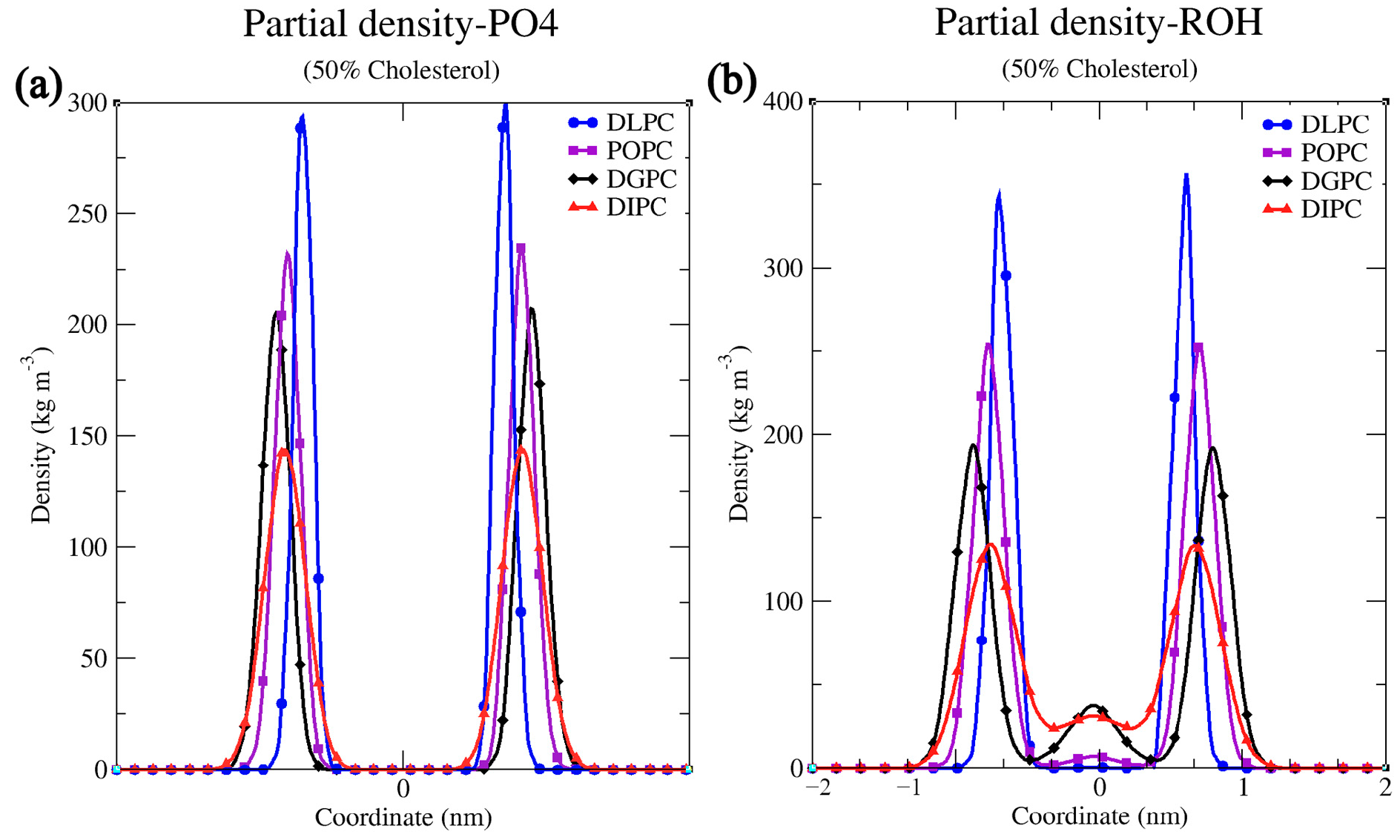
Coarse-Grained Molecular Dynamics (MD) simulations were conducted for various phosphatidylcholine (PC) lipids, including DLPC, POPC, DGPC, and DIPC, at different cholesterol concentrations. Bilayer thickness, order parameter, and area per lipid were analyzed using the simulation trajectory files. Fluctuations in these bilayer parameters indicate that sterols have a major impact on the bilayer’s physical structure with the increasing and decreasing concentration. Other lipids were chosen based on their chemical structure i.e., the presence of their double bond position and number of double bonds present in their tails. Subsequent simulations were performed with similar Cholesterol concentrations across the bilayer which resulted in the more significant observation in their Bilayer Thickness, Order Parameter, and Radial Distribution Function (RDF) study. The results showed the importance of double bonds as it plays a crucial role in the movement of sterols inside the bilayer where more ordered behavior is observed for DLPC and a decrease in thickness and order parameters for DIPC. A cartoon representation of this phenomenon is shown in Figure 10. Furthermore, lipid saturation is directly affected by the number of double bonds present in the hydrocarbon chains. To confirm this, the lipids like POPC and DGPC were simulated under the same condition. This confirmed to us the structural properties of the lipid’s acyl chain play a significant role in the movement and compartmentalization of molecules inside the bilayer. Further understanding of cholesterol’s role in modulating lipid bilayer phase behavior offers valuable insights for future experimental studies, particularly in health-related research, drug delivery, and membrane design [58,59]. Cholesterol’s ability to fine-tune the balance between ordered and disordered phases impacts membrane fluidity, permeability, and protein function, influencing conditions such as cardiovascular disease, Alzheimer’s, and cancer [60,61,62]. This knowledge can guide the development of targeted drug delivery systems, design biomimetic membranes that replicate natural properties, and study protein-membrane interactions [63]. Additionally, it provides a basis for investigating membrane heterogeneity, phase separation, and temperature-dependent transitions, advancing our understanding of cellular processes and therapeutic strategies [64,65,66].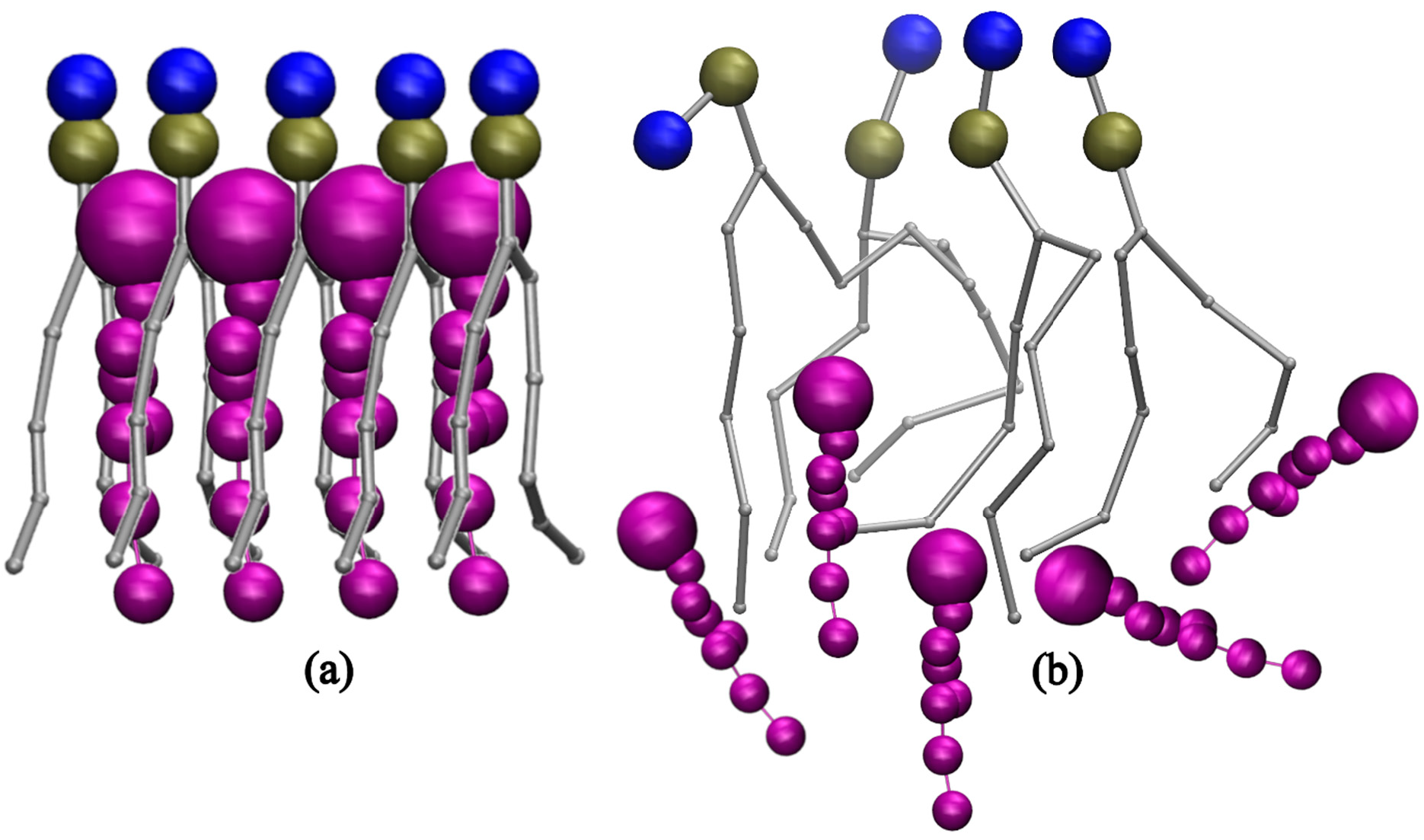
| .gro | Gromacs File |
| .itp | Portable Topology File |
| .top | Topology File |
| .xtc | Trajectory File |
| APL | Area per Lipid |
| BT | Bilayer Thickness |
| Chol | Cholesterol |
| DGPC | 1,2-di-(11Z-eicosenoyl)-sn-glycero-3-phosphocholine |
| DIPC | 1,2-Dilinoleoyl-sn-glycero-3-phosphocholine |
| DLPC | 1,2-Dilauroyl-rac-glycero-3-phosphocholine |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine |
| DOPC | Dioleoylphosphatidylcholine |
| DPPC | Dipalmitoylphosphatidylcholine |
| FATSlim | Fast Analysis Toolbox for Simulation of Lipid Membranes |
| GROMACS | GROningen MAchine for Chemical Simulations |
| INSANE | INSert membrANE |
| LINCS | LINear Constraint Solver |
| MPI | Message Passing Interface |
| PC | Phosphatidylcholine |
| POPC | 1-Palmitoyl-2-oleoylphosphatidylcholine |
| POPS | 1,2-Palmitoyl-oleoyl-sn-glycero-3-phosphoserine |
| RDF | Radial Distribution Function |
| VMD | Visual Molecular Dynamics |
Conceptualization and supervision, A.V.R.M.; methodology, D.B.; validation, A.V.R.M.; formal analysis, D.B.; investigation, D.B.; resources, A.V.R.M.; data curation. D.B.; writing—original draft preparation, D.B.; writing—review and editing, D.B. and A.V.R.M; project administration, A.V.R.M.; funding acquisition, A.V.R.M. All authors have read and agreed to the published version of the manuscript.
Data supporting the results of this study are available upon request from the corresponding author.
No consent for publication is required, as the manuscript does not involve any individual personal data, images, videos, or other materials that would necessitate consent.
The authors declare that they have no conflicts of interest, financial or personal, that could have influenced the work reported in this manuscript.
This work received a grant from the Defence Institute of Advanced Technology (DIAT) with the grant number [DIAT/F/Adm/Project/OM/Corr/P-49].
The authors would like to acknowledge DIAT for funding and infrastructural support.
[1] Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpi, J.L. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [CrossRef]
[2] Allen, M.-P. Introduction to Molecular Dynamics Simulation. 2004 Available online: https://juser.fz-juelich.de/record/152581/files/FZJ-2014-02193.pdf (accessed on 17 July 2024).
[3] Lindahl, E.; Sansom, M. Membrane Proteins: Molecular Dynamics Simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [CrossRef] [PubMed]
[4] Feller, S.E. Molecular Dynamics Simulations of Lipid Bilayers. Curr. Opin. Colloid Interface Sci. 2000, 5, 217–223. [CrossRef]
[5] Vigh, L.; Escriba, P.V.; Sonnleitner, A.; Sonnleitner, M.; Piotto, S.; Maresca, B.; Horváth, I.; Harwood, J.L. The Significance of Lipid Composition for Membrane Activity: New Concepts and Ways of Assessing Function. Prog. Lipid Res. 2005, 44, 303–344. [CrossRef] [PubMed]
[6] Burke, K.A.; Yates, E.A.; Legleiter, J. Biophysical Insights into How Surfaces, Including Lipid Membranes, Modulate Protein Aggregation Related to Neurodegeneration. Front. Neurol. 2013, 4. [CrossRef] [PubMed]
[7] Moradi, S.; Nowroozi, A.; Shahlaei, M. Shedding Light on the Structural Properties of Lipid Bilayers Using Molecular Dynamics Simulation: A Review Study. RSC Adv. 2019, 9, 4644–4658. [CrossRef]
[8] Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [CrossRef]
[9] Marrink, S.J.; Berendsen, H.J.C. Permeation Process of Small Molecules Across Lipid Membranes Studied by Molecular Dynamics Simulations. J. Phys. Chem. 1996, 100, 16729–16738. [CrossRef]
[10] Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [CrossRef]
[11] Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [CrossRef]
[12] Buchoux, S. FATSLiM: A Fast and Robust Software to Analyze MD Simulations of Membranes. Bioinformatics 2017, 33, 133–134. [CrossRef] [PubMed]
[13] Allen, W.J.; Lemkul, J.A.; Bevan, D.R. GridMAT-MD: A Grid-Based Membrane Analysis Tool for Use with Molecular Dynamics. J. Comput. Chem. 2009, 30, 1952–1958. [CrossRef] [PubMed]
[14] Arora, A.; Gupta, C.M. Glycerol Backbone Conformation in Phosphatidylcholines Is Primarily Determined by the Intramolecular Stacking of the Vicinally Arranged Acyl chains. Biochim. Biophys. Acta (BBA)-Biomembr. 1997, 1324, 47–60. [CrossRef]
[15] Yèagle, P.L. Lipid Regulation of Cell Membrane Structure and Function. FASEB J. 1989, 3, 1833–1842. [CrossRef]
[16] Robertson, J.L. The Lipid Bilayer Membrane and Its Protein Constituents. J. Gen. Physiol. 2018, 150, 1472–1483. [CrossRef] [PubMed]
[17] Hagve, T.-A. Effects of Unsaturated Fatty Acids on Cell Membrane Functions. Scand. J. Clin. Lab. Investig. 1988, 48, 381–388. [CrossRef]
[18] Ghosh, S.; Strum, J.C.; Bell, R.M. Lipid Biochemistry: Functions of Glycerolipids and Sphingolipids in Cellular Signaling. FASEB J. 1997, 11, 45–50. [CrossRef]
[19] Dennis, E.A.; Rhee, S.G.; Billah, M.M.; Hannun, Y.A. Role of Phospholipases in Generating Lipid Second Messengers in Signal Transduction. FASEB J. 1991, 5, 2068–2077. [CrossRef]
[20] Sanchez-Alvarez, M.; Zhang, Q.; Finger, F.; Wakelam, M.J.O.; Bakal, C. Cell Cycle Progression Is an Essential Regulatory Component of Phospholipid Metabolism and Membrane Homeostasis. Open Biol. 2015, 5. [CrossRef]
[21] . In Intravenous Lipid Emulsions Volume 112, Calder, P.C.; Waitzberg, D.L.; Koletzko, B., Eds.; S. Karger AG: Basel, Switzerland, 2014; . . [CrossRef]
[22] Shrestha, H.; Bala, R.; Arora, S. Lipid-Based Drug Delivery Systems. J. Pharm. 2014, 2014, 1–10. [CrossRef]
[23] Weiner, N.; Martin, F.; Riaz, M. Liposomes as a Drug Delivery System. Drug Dev. Ind. Pharm. 1989, 15, 1523–1554. [CrossRef]
[24] Law, S.-H.; Chan, M.-L.; Marathe, G.K.; Parveen, F.; Chen, C.-H.; Ke, L.-Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20. [CrossRef]
[25] Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21. [CrossRef] [PubMed]
[26] Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in Cardiovascular Diseases. Redox Biol. 2019, 23, 101119. [CrossRef] [PubMed]
[27] Paltauf, F.; Hermetter, A. Phospholipids—Natural, Semisynthetic, Synthetic. Phospholipids ; Springer: Boston, MA, USA, 1990; 1–12. . [CrossRef]
[28] Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid–Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [CrossRef] [PubMed]
[29] Corey, R.A.; Stansfeld, P.J.; Sansom, M.S. The energetics of protein–lipid interactions as viewed by molecular simulations. Biochem. Soc. Trans. 2019, 48, 25–37. [CrossRef]
[30] Bloch, K.E. Sterol, Structure and Membrane Function. Crit. Rev. Biochem. 1983, 14, 47–92. [CrossRef]
[31] Dufourc, E.J. Sterols and Membrane Dynamics. J. Chem. Biol. 2008, 1, 63–77. [CrossRef]
[32] Edholm, O.; Nyberg, A. Cholesterol in Model Membranes. A Molecular Dynamics Simulation. Biophys. J. 1992, 63, 1081–1089. [CrossRef]
[33] Czub, J.; Baginski, M. Comparative Molecular Dynamics Study of Lipid Membranes Containing Cholesterol and Ergosterol. Biophys. J. 2006, 90, 2368–2382. [CrossRef]
[34] Kamble, S.; Patil, S.; Appala, V.R.M. Nano-Mechanical Characterization of Asymmetric DLPC/DSPC Supported Lipid Bilayers. Chem. Phys. Lipids 2021, 234, 105007. [CrossRef] [PubMed]
[35] Kamble, S.; Patil, S.; Kulkarni, M.; Appala, V.R.M. Interleaflet Decoupling in a Lipid Bilayer at Excess Cholesterol Probed by Spectroscopic Ellipsometry and Simulations. J. Membr. Biol. 2020, 253, 647–659. [CrossRef] [PubMed]
[36] Murthy, A.V.R.; Guyomarc’H, F.; Paboeuf, G.; Vié, V.; Lopez, C. Cholesterol Strongly Affects the Organization of Lipid Monolayers Studied as Models of the Milk Fat Globule Membrane: Condensing Effect and Change in the Lipid Domain Morphology. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2308–2316. [CrossRef] [PubMed]
[37] Murthy, A.V.R.; Guyomarc’H, F.; Lopez, C. The Temperature-Dependent Physical State of Polar Lipids and Their Miscibility Impact the Topography and Mechanical Properties of Bilayer Models of the Milk Fat Globule Membrane. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 2181–2190. [CrossRef]
[38] Chiu, S.; Jakobsson, E.; Mashl, R.J.; Scott, H.L. Cholesterol-Induced Modifications in Lipid Bilayers: A Simulation Study. Biophys. J. 2002, 83, 1842–1853. [CrossRef]
[39] Robinson, A.; Richards, W.; Thomas, P.; Hann, M. Behavior of Cholesterol and Its Effect on Head Group and Chain Conformations in Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 1995, 68, 164–170. [CrossRef]
[40] Snyder, R.G.; Tu, K.; Klein, M.L.; Mendelssohn, R.; Strauss, H.L.; Sun, W. Acyl Chain Conformation and Packing in Dipalmitoylphosphatidylcholine Bilayers from MD Simulation and IR Spectroscopy. J. Phys. Chem. B 2002, 106, 6273–6288. [CrossRef]
[41] Piggot, T.J.; Allison, J.R.; Sessions, R.B.; Essex, J.W. On the Calculation of Acyl Chain Order Parameters from Lipid Simulations. J. Chem. Theory Comput. 2017, 13, 5683–5696. [CrossRef]
[42] Vermeer, L.S.; de Groot, B.L.; Réat, V.; Milon, A.; Czaplicki, J. Acyl Chain Order Parameter Profiles in Phospholipid Bilayers: Computation from Molecular Dynamics Simulations and Comparison with 2H NMR Experiments. Eur. Biophys. J. 2007, 36, 919–931. [CrossRef]
[43] Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A General Purpose Force Field for Coarse-Grained Molecular Dynamics. Nat. Methods 2021, 18, 382–388. [CrossRef]
[44] Kamble, S.; Patil, S.; Kulkarni, M.; Murthy, A.V.R. Spectroscopic Ellipsometry of Fluid and Gel Phase Lipid Bilayers in Hydrated Conditions. Colloids Surfaces B Biointerfaces 2018, 176, 55–61. [CrossRef]
[45] Adhyapak, P.R.; Panchal, S.V.; Murthy, A.V.R. Cholesterol Induced Asymmetry in DOPC Bilayers Probed by AFM Force Spectroscopy. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 953–959. [CrossRef]
[46] Pantelopulos, G.A.; Straub, J.E. Regimes of Complex Lipid Bilayer Phases Induced by Cholesterol Concentration in MD Simulation. Biophys. J. 2018, 115, 2167–2178. [CrossRef] [PubMed]
[47] Ermilova, I.; Lyubartsev, A.P. Cholesterol in Phospholipid Bilayers: Positions and Orientations Inside Membranes with Different Unsaturation Degrees. Soft Matter 2019, 15, 78–93. [CrossRef] [PubMed]
[48] Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical Properties of Model Biological Lipid Bilayers: Insights from All-Atom Molecular Dynamics Simulations. J. Mol. Model. 2019, 25, 76. [CrossRef]
[49] Khakbaz, P.; Klauda, J.B. Investigation of Phase Transitions of Saturated Phosphocholine Lipid Bilayers via Molecular Dynamics Simulations. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 1489–1501. [CrossRef]
[50] Chakraborty, S.; Doktorova, M.; Molugu, T.R.; Heberle, F.A.; Scott, H.L.; Dzikovski, B.; Nagao, M.; Stingaciu, L.-R.; Standaert, R.F.; Barrera, F.N.; et al. How Cholesterol Stiffens Unsaturated Lipid Membranes. Proc. Natl. Acad. Sci. USA 2020, 117, 21896–21905. [CrossRef]
[51] Dutta, A.; Kumari, M.; Kashyap, H.K. Tracking Cholesterol Flip-Flop in Mammalian Plasma Membrane through Coarse-Grained Molecular Dynamics Simulations. Langmuir 2025, 41, 1651–1663. [CrossRef] [PubMed]
[52] Zhang, Y.; Lervik, A.; Seddon, J.; Bresme, F. A Coarse-Grained Molecular Dynamics Investigation of the Phase Behavior of DPPC/Cholesterol Mixtures. Chem. Phys. Lipids 2015, 185, 88–98. [CrossRef]
[53] Kučerka, N.; Perlmutter, J.D.; Pan, J.; Tristram-Nagle, S.; Katsaras, J.; Sachs, J.N. The Effect of Cholesterol on Short- and Long-Chain Monounsaturated Lipid Bilayers as Determined by Molecular Dynamics Simulations and X-Ray Scattering. Biophys. J. 2008, 95, 2792–2805. [CrossRef]
[54] Alwarawrah, M.; Dai, J.; Huang, J. A Molecular View of the Cholesterol Condensing Effect in DOPC Lipid Bilayers. J. Phys. Chem. B 2010, 114, 7516–7523. [CrossRef] [PubMed]
[55] Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [CrossRef]
[56] Limonada—Home. Available online: https://limonada.univ-reims.fr/ (accessed on 17 July 2024).
[57] Doole, F.T.; Kumarage, T.; Ashkar, R.; Brown, M.F. Cholesterol Stiffening of Lipid Membranes. J. Membr. Biol. 2022, 255, 385–405. [CrossRef] [PubMed]
[58] Zhang, Y.; Zhang, T.; Liang, Y.; Jiang, L.; Sui, X. Dietary Bioactive Lipids: A Review on Absorption, Metabolism, and Health Properties. J. Agric. Food Chem. 2021, 69, 8929–8943. [CrossRef]
[59] Matei, A.-M.; Caruntu, C.; Tampa, M.; Georgescu, S.R.; Matei, C.; Constantin, M.M.; Constantin, T.V.; Calina, D.; Ciubotaru, D.A.; Badarau, I.A.; et al. Applications of Nanosized-Lipid-Based Drug Delivery Systems in Wound Care. Appl. Sci. 2021, 11. [CrossRef]
[60] Marzoog, B.A.; Vlasova, T.I. Membrane Lipids Under Norm and Pathology. Eur. J. Clin. Exp. Med. 2021, 19, 59–75. [CrossRef]
[61] Torres, M.; Parets, S.; Fernández-Díaz, J.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Román, R.; Lladó, V.; Rosselló, C.A.; Fernández-García, P.; Escribá, P.V. Lipids in Pathophysiology and Development of the Membrane Lipid Therapy: New Bioactive Lipids. Membranes 2021, 11. [CrossRef]
[62] Rudajev, V.; Novotny, J. Cholesterol as a Key Player in Amyloid β-Mediated Toxicity in Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15. [CrossRef] [PubMed]
[63] Sarkis, J.; Vié, V. Biomimetic Models to Investigate Membrane Biophysics Affecting Lipid–Protein Interaction. Front. Bioeng. Biotechnol. 2020, 8. [CrossRef]
[64] Walde, P.; Ichikawa, S. Lipid Vesicles and Other Polymolecular Aggregates—From Basic Studies of Polar Lipids to Innovative Applications. Appl. Sci. 2021, 11. [CrossRef]
[65] Serdiuk, T.; Manna, M.; Zhang, C.; Mari, S.A.; Kulig, W.; Pluhackova, K.; Kobilka, B.K.; Vattulainen, I.; Müller, D.J. A Cholesterol Analog Stabilizes the Human β2 -Adrenergic Receptor Nonlinearly with Temperature. Sci. Signal. 2022, 15, eabi7031. [CrossRef] [PubMed]
[66] Musumeci, T.; Bonaccorso, A.; Carbone, C. Basic Concepts of Liposomes. Liposomes in Drug Delivery ; Elsevier: Amsterdam, The Netherlands, 2024; 19–48. . [CrossRef]