
 ACCESS
Research Article
ACCESS
Research Article
Volume 2, Article ID: 2026.0012

Natalya Risinskaya
risinska@gmail.com


Sofia Starchenko
starsof1309@mail.ru


Yulia Chabaeva
uchabaeva@gmail.com


Abdulpatakh Abdulpatakhov
patakh1997@mail.ru


Ekaterina Kotova
kotova.e@blood.ru


Valeriya Surimova
surimova.lera@mail.ru


Dmitry Bessmertnyy
dmitry_bessmertnyy@mail.ru


Anastasia Kashlakova
kashlakova.a@blood.ru


Anastasia Vasileva
vasilnastia@yandex.ru


Zalina Fidarova
fidarova.z@blood.ru


Olga Aleshina
gavrilina.o@blood.ru


Anna Yushkova
ann.unikova@bk.ru


Olga Dubova
doe30102001@gmail.com


Kseniya Nikiforova
nikiforova.k@blood.ru


Nikolay Kapranov
kapranov.n@blood.ru


Irina Galtseva
galtseva.i@blood.ru


Alina Ponomareva
a.ponomareva@genomed.ru


Ilya Kanivets
dr.kanivets@genomed.ru,


Sergey Korostelev
korostelevsa@genomed.ru


Sergey Kulikov
smkulikov@mail.ru


Elena Parovichnikova
parovichnikova.e@blood.ru

1 National Medical Research Center for Hematology, 125167 Moscow, Russia
2 Genomed Laboratory of Molecular Pathology, 115419 Moscow, Russia
* Author to whom correspondence should be addressed
Received: 02 Dec 2025 Accepted: 27 Jan 2026 Available Online: 02 Feb 2026
Background: The tumor suppressor p53 is a critical regulator of gene expression in cancer, acting through binding to specific p53 response elements (REs). One of key RE rs4590952 is found in the KITLG gene.
Aims: This study aimed to precisely investigate the allelic variants of the SNP rs4590952 within patient groups with acute leukemia and to analyze their association with the disease and response to therapy. A specific objective was to determine the frequency and nature of regions of homozygosity encompassing the KITLG locus.
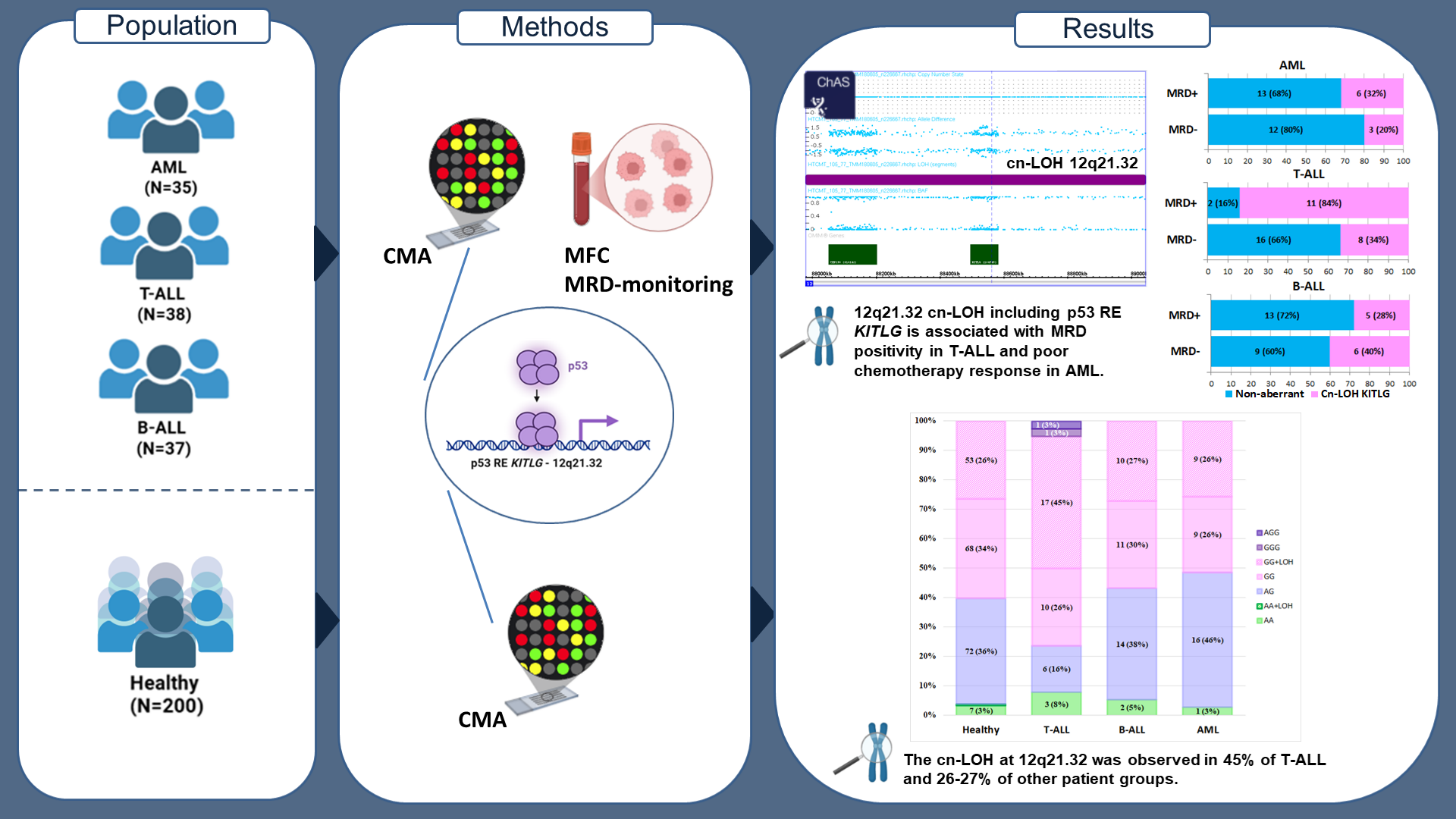
Methods: The study cohort included: 37 patients with Ph-negative B-ALL, 38 with T-ALL, and 35 with de novo intermediate-risk AML. A reference group of 200 healthy individuals without oncohematological disorders was used for comparison. Chromosomal microarray analysis (CMA) was performed using the CytoScan™ HT-CMA. Statistical analysis was performed using Python 3.12.4 and SAS 9.4.
Results: The allele frequency (AF) of the G allele of rs4590952 was 0.785 in the reference group. Among patients, the AF was 0.833 in T-ALL, 0.757 in B-ALL, and 0.743 in AML. The frequency of copy-neutral loss of heterozygosity (cn-LOH) at 12q21.32, which results in the GG genotype, was significantly higher in the T-ALL group (45%) compared to the reference group (27%) (OR=0.4; 95% CI: 0.2–0.9; p=0.02). A significant association between MRD-positive status and cn-LOH KITLG was found specifically in the T-ALL group (OR=11; 95% CI: 2–62; p=0.005). Cn-LOH KITLG was also significantly associated with poor chemotherapy response in AML (p=0.01).
Summary: The GG genotype of the p53 response element in KITLG (rs4590952) frequently arises from acquired cn-LOH at 12q21.32, observed in 45% of T-ALL and 26-27% of other cases. This treatment-response marker, present irrespective of leukemic status, warrants further validation in expanded cohorts.
Disclaimer: This is not the final version of the article. Changes may occur when the manuscript is published in its final format.