
 ACCESS
Review Article
ACCESS
Review Article
Volume 2, Article ID: 2025.0020

Komal Parmar
komal.parmar2385@gmail.com


Rajendra Patel
raju1857@yahoo.co.in

1 ROFEL Shri G.M. Bilakhia College of Pharmacy, Rajju Shroff ROFEL University, Vapi, India
2 Department of Pharmaceutical Sciences, Gujarat Technological University, Ahmedabad, India
* Author to whom correspondence should be addressed
Received: 16 Jul 2025 Accepted: 15 Oct 2025 Available Online: 21 Oct 2025
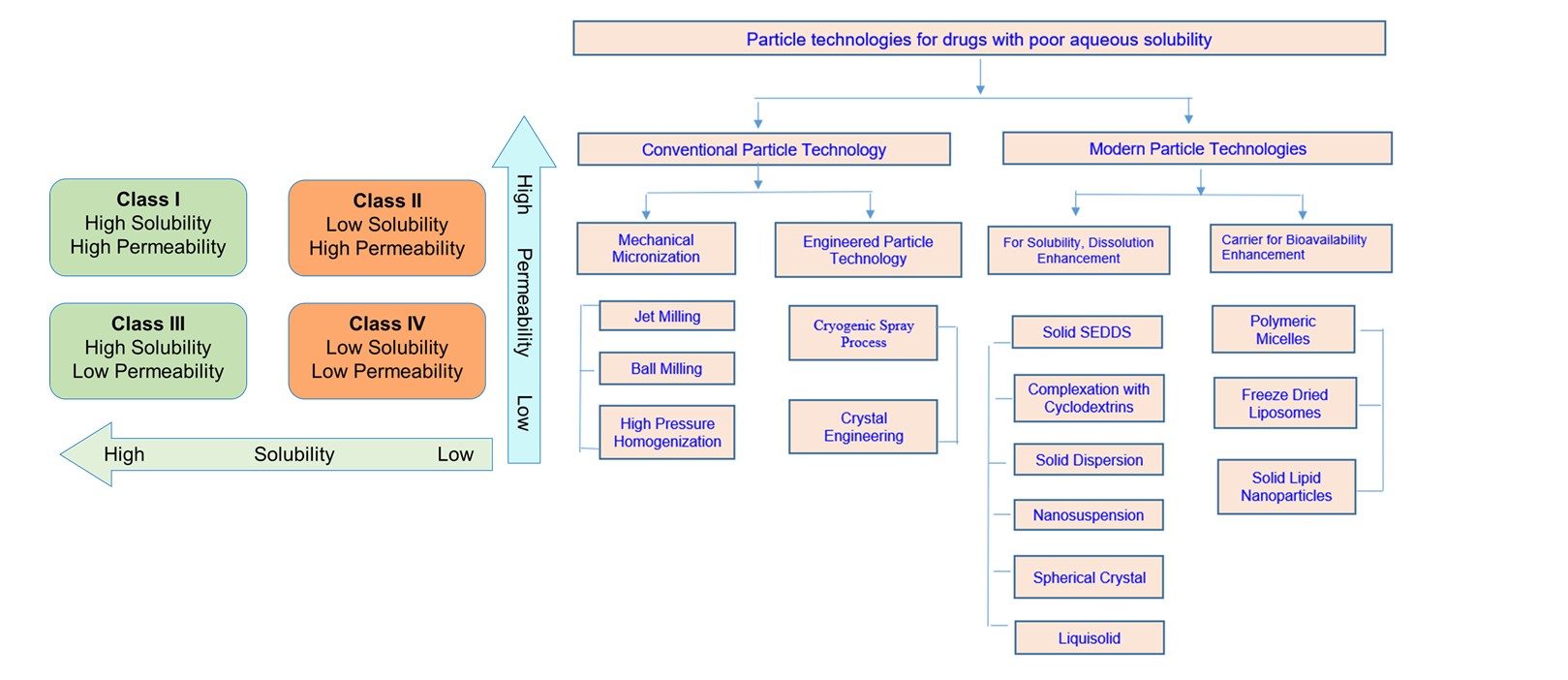
For any drug to be therapeutically efficacious it must enter the systemic circulation and in order to do so, the administered drug must be dissolved in the GIT. Approximately 40 % of drugs developed in the past and about 70-90 % of drugs in development were found to be poorly water soluble. Various pharmaceutical particle technologies are applied to enhance the aqueous solubility of poorly soluble drugs that restrict in vivo bioavailability upon oral administration due to their low dissolution rate in gastrointestinal fluids. The approach involves from traditional to modern particle technologies such as micronization, complexation, nano-suspension, and others. The employed technologies modify the drug's solubility properties, produce drug forms that are readily soluble in water and can be easily formulated into different dosage forms. The aim of this review paper is to summarize the key aspects of currently used particle technologies to enhance the solubility, dissolution and bioavailability of poorly water-soluble drugs.
Disclaimer : This is not the final version of the article. Changes may occur when the manuscript is published in its final format.